- Наследственные пигментные гепатозы. Синдром Жильбера, Криглера-Найяра, Дабина-Джонсона,-Ротора

Содержание
- 2. Наследственные пигментные гепатозы Наследственные пигментные гепатозы – группа заболеваний с доброкачественным течением, связанных с нарушением внутрипеченочного
- 3. определение Е80.4. Синдром Жильбера.(Аутосомно-доминантный тип наследования) Синдром Жильбера - пигментный гепатоз (простая семейная холемия, конституциональная гипербилирубинемия,
- 4. Этиология и патогенез Синдром обусловлен мутацией в гене UGT1A1, который кодирует фермент уридиндифосфатглюкуронилтрансферазу (УДФГТ). В патогенезе
- 5. Обмен билирубина складывается из транспорта его в плазме крови, захвата печенью, конъюгации, билиарной экскреции (рис. 6-1).
- 6. цитов, захват его внутрипеченочным протеином. Конъюгацию билирубина с образованием моно- и диглюкуронидов (конъюгированного билирубина) обеспечивает УДФГТ.
- 7. Рис. 6-1. Обмен и конъюгирование билирубина
- 8. жалобы Больные часто могут жаловаться на тяжесть, постоянные тупые боли в правом подреберье, тошноту, рвоту, отсутствие
- 9. Клиническая картина Заболевание протекает триадой симптомов: перемежающаяся желтуха – гипербилирубиномия обусловлена преимущественно повышением уровня неконъюгированного билирубина;
- 10. Лабораторная диагностика Диагностика заболевания подразумевает проведение тестов. Тест на содержание билирубина в сыворотке крови, которое повышается
- 11. Инструментальная диагностика Молекулярная диагностика включает анализ ДНК гена УДФГТ. С помощью УЗИ органов брюшной полости определяют
- 12. Синдром Жильбера: а - пациентка - участница конкурса красоты; б - УЗИ: изменения отсутствуют; в -
- 13. определение E80.5. Синдром Криглера-Найяра. Синдром Криглера Найяра - врожденная неконъюгированная гипербилирубинемия с аутосомно-доминантным (I типом синдрома
- 14. Этиология и патогенез Гипербилирубинемия является следствием нарушения конъюгации в печени билирубина с глюкуроновой кислотой, обусловленного отсутствием
- 15. Клиническая картина Характерны симптомы желтухи (желтушность склер и кожного покрова) и неврологические нарушения (билирубиновая энцефалопатия). I
- 16. II тип занимает промежуточное положение по тяжести клинических проявлений между синдромом Криглера- Найяра I типа и
- 17. Диагностика При I типе синдрома Криглера-Найяра основной биохимический показатель - уровень билирубина в крови выше 200
- 18. Дифференциальная диагностика различных типов желтухи и синдрома Криглера-Найяра I и II типа
- 19. определение Синдром Дабина-Джонсона - энзимопатическая желтуха, редкий пигментный гепатоз, характеризуемый нарушением экскреции связанного билирубина из гепатоцитов
- 20. Этиология и патогенез Заболевание обусловлено наследственным дефектом с аутосомно-рецессивным типом наследования. Генетический дефект заключается в появлении
- 21. Жалобы Пациенты жалуются на повышенную утомляемость, плохой аппетит, боль в правом подреберье. Выраженность желтухи может варьировать,
- 22. Диагностика При биохимическом исследовании крови определяется преобладание фракции прямого билирубина, в связи с этим имеется и
- 23. Патоморфология Синдром Дабина-Джонсона: а - макропрепарат: «шоколадная печень»; б - скопление желчи и темного пигмента (окраска
- 24. определение E80.6. Синдром Ротора. Синдром Ротора - наследственный пигментный гепатоз с аутосомно-рецессивным типом наследования, напоминающий синдром
- 25. Этиология и патогенез В основе заболевания лежит генетический дефект, наследуемый по аутосомно-рецессивному типу. Патогенез связан не
- 26. клиника Заболевание манифестирует в детском возрасте и проявляется эпизодами нерезко выраженной желтухи. В некоторых случаях наблюдаются
- 27. Дифференциальная диагностика
- 29. Скачать презентацию

Наследственные пигментные гепатозы
Наследственные пигментные гепатозы – группа заболеваний с доброкачественным течением,
Наследственные пигментные гепатозы
Наследственные пигментные гепатозы – группа заболеваний с доброкачественным течением,

определение
Е80.4. Синдром Жильбера.(Аутосомно-доминантный тип наследования)
Синдром Жильбера - пигментный гепатоз (простая семейная
определение
Е80.4. Синдром Жильбера.(Аутосомно-доминантный тип наследования)
Синдром Жильбера - пигментный гепатоз (простая семейная

Этиология и патогенез
Синдром обусловлен мутацией в гене UGT1A1, который кодирует фермент уридиндифосфатглюкуронилтрансферазу (УДФГТ).
Этиология и патогенез
Синдром обусловлен мутацией в гене UGT1A1, который кодирует фермент уридиндифосфатглюкуронилтрансферазу (УДФГТ).

Обмен билирубина складывается из транспорта его в плазме крови, захвата печенью, конъюгации,
Обмен билирубина складывается из транспорта его в плазме крови, захвата печенью, конъюгации,

цитов, захват его внутрипеченочным протеином. Конъюгацию билирубина с образованием моно- и
цитов, захват его внутрипеченочным протеином. Конъюгацию билирубина с образованием моно- и

Рис. 6-1.
Обмен и конъюгирование билирубина
Рис. 6-1.
Обмен и конъюгирование билирубина

жалобы
Больные часто могут жаловаться на тяжесть, постоянные тупые боли в правом
жалобы
Больные часто могут жаловаться на тяжесть, постоянные тупые боли в правом

Клиническая картина
Заболевание протекает триадой симптомов:
перемежающаяся желтуха – гипербилирубиномия обусловлена преимущественно повышением
Клиническая картина
Заболевание протекает триадой симптомов:
перемежающаяся желтуха – гипербилирубиномия обусловлена преимущественно повышением

Лабораторная диагностика
Диагностика заболевания подразумевает проведение тестов.
Тест на содержание билирубина в сыворотке
Лабораторная диагностика
Диагностика заболевания подразумевает проведение тестов.
Тест на содержание билирубина в сыворотке

Инструментальная диагностика
Молекулярная диагностика включает анализ ДНК гена УДФГТ.
С помощью УЗИ органов
Инструментальная диагностика
Молекулярная диагностика включает анализ ДНК гена УДФГТ.
С помощью УЗИ органов
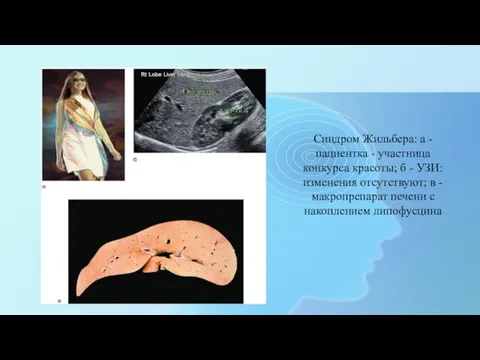
Синдром Жильбера: а - пациентка - участница конкурса красоты; б -
Синдром Жильбера: а - пациентка - участница конкурса красоты; б -

определение
E80.5. Синдром Криглера-Найяра.
Синдром Криглера Найяра - врожденная неконъюгированная гипербилирубинемия с аутосомно-доминантным
определение
E80.5. Синдром Криглера-Найяра.
Синдром Криглера Найяра - врожденная неконъюгированная гипербилирубинемия с аутосомно-доминантным

Этиология и патогенез
Гипербилирубинемия является следствием нарушения конъюгации в печени билирубина с
Этиология и патогенез
Гипербилирубинемия является следствием нарушения конъюгации в печени билирубина с

Клиническая картина
Характерны симптомы желтухи (желтушность склер и кожного покрова) и неврологические
Клиническая картина
Характерны симптомы желтухи (желтушность склер и кожного покрова) и неврологические

II тип занимает промежуточное положение по тяжести клинических проявлений между синдромом Криглера-
Найяра I
II тип занимает промежуточное положение по тяжести клинических проявлений между синдромом Криглера-
Найяра I

Диагностика
При I типе синдрома Криглера-Найяра основной биохимический показатель - уровень билирубина в крови
Диагностика
При I типе синдрома Криглера-Найяра основной биохимический показатель - уровень билирубина в крови

Дифференциальная диагностика различных типов желтухи и синдрома Криглера-Найяра I и II
Дифференциальная диагностика различных типов желтухи и синдрома Криглера-Найяра I и II

определение
Синдром Дабина-Джонсона - энзимопатическая желтуха, редкий пигментный гепатоз, характеризуемый нарушением экскреции
определение
Синдром Дабина-Джонсона - энзимопатическая желтуха, редкий пигментный гепатоз, характеризуемый нарушением экскреции

Этиология и патогенез
Заболевание обусловлено наследственным дефектом с аутосомно-рецессивным типом наследования. Генетический
Этиология и патогенез
Заболевание обусловлено наследственным дефектом с аутосомно-рецессивным типом наследования. Генетический

Жалобы
Пациенты жалуются на повышенную утомляемость, плохой аппетит, боль в правом подреберье.
Жалобы
Пациенты жалуются на повышенную утомляемость, плохой аппетит, боль в правом подреберье.

Диагностика
При биохимическом исследовании крови определяется преобладание фракции прямого билирубина, в связи
Диагностика
При биохимическом исследовании крови определяется преобладание фракции прямого билирубина, в связи

Патоморфология
Синдром Дабина-Джонсона: а - макропрепарат: «шоколадная печень»; б - скопление желчи
Патоморфология Синдром Дабина-Джонсона: а - макропрепарат: «шоколадная печень»; б - скопление желчи

определение
E80.6. Синдром Ротора.
Синдром Ротора - наследственный пигментный гепатоз с аутосомно-рецессивным типом
определение
E80.6. Синдром Ротора.
Синдром Ротора - наследственный пигментный гепатоз с аутосомно-рецессивным типом

Этиология и патогенез
В основе заболевания лежит генетический дефект, наследуемый по аутосомно-рецессивному
Этиология и патогенез
В основе заболевания лежит генетический дефект, наследуемый по аутосомно-рецессивному

клиника
Заболевание манифестирует в детском возрасте и проявляется эпизодами нерезко выраженной желтухи.
клиника
Заболевание манифестирует в детском возрасте и проявляется эпизодами нерезко выраженной желтухи.

Дифференциальная диагностика
Дифференциальная диагностика
 Ролевые и деловые игры
Ролевые и деловые игры Клінічні випадки Завдання
Клінічні випадки Завдання Үй жұмысын тексеру. Есеп шығару
Үй жұмысын тексеру. Есеп шығару Инновационные технологии в ортодонтии
Инновационные технологии в ортодонтии Профессиональная деятельность медицинской сестры при черепно-мозговой травме
Профессиональная деятельность медицинской сестры при черепно-мозговой травме Осложнения анестезии
Осложнения анестезии Мужское здоровье
Мужское здоровье История развития психопатологии в зарубежный странах
История развития психопатологии в зарубежный странах Новый порядок проведения профилактического осмотра и диспансеризации в рамках Приказа Министерства здравоохранения РФ №124н
Новый порядок проведения профилактического осмотра и диспансеризации в рамках Приказа Министерства здравоохранения РФ №124н Основы эпидемиологии и иммунологии. Меры профилактики инфекционных заболеваний
Основы эпидемиологии и иммунологии. Меры профилактики инфекционных заболеваний Выделительная система
Выделительная система Жүрекің тәждік (коронарлық) жетіспеушілігінің диагностикасында коронарографияның, аортографияның, вентрикулографияның
Жүрекің тәждік (коронарлық) жетіспеушілігінің диагностикасында коронарографияның, аортографияның, вентрикулографияның Технология приготовления липосомальных форм лекарственных препаратов и их применение
Технология приготовления липосомальных форм лекарственных препаратов и их применение Движение без боли. Здоровье и молодость вместе с Devita
Движение без боли. Здоровье и молодость вместе с Devita Сочетание речевых средств и невербальных действий
Сочетание речевых средств и невербальных действий Туберкулез костей и суставов
Туберкулез костей и суставов Острый перитонит
Острый перитонит Потребностно-мотивационная сфера личности
Потребностно-мотивационная сфера личности Современные аспекты интенсивного лечения пострадавших с политравмой
Современные аспекты интенсивного лечения пострадавших с политравмой Психология преступных групп
Психология преступных групп Основные аспекты взаимодействия пищи и ЛС
Основные аспекты взаимодействия пищи и ЛС Сущность биосоциальной природы психики и поведения человека
Сущность биосоциальной природы психики и поведения человека Инфекционный процесс. Сепсис. ВИЧ - инфекция
Инфекционный процесс. Сепсис. ВИЧ - инфекция Структура приемного покоя детского стационара
Структура приемного покоя детского стационара Выявление туберкулёза согласно стратегии ДОТС
Выявление туберкулёза согласно стратегии ДОТС Общие правила транспортировки пострадавшего
Общие правила транспортировки пострадавшего Обзор эрготерапии
Обзор эрготерапии Наркотические анальгетики
Наркотические анальгетики