- Виды мутаций

Содержание
- 2. Причины мутаций Физические факторы (различные виды ионизирующей радиации, ультрафиолетовое излучение) Химические факторы (инсектициды, гербициды, наркотики, алкоголь,
- 3. Наследственные заболевания заболевания человека, обусловленные хромосомными и генными мутациями, передаваемыми по наследству через гаметы.
- 4. Историческая справка В 1929 г. советский генетик, невропатолог С.Н.Давиденко организовал первую в мире медико-генетическую консультацию. Он
- 5. Классификация наследственных болезней
- 6. Таблица для заполнения Д\з
- 7. Генные заболевания Аутосомно-доминирующий тип наследования 1. Болезнь встечается в каждом поколении родословной. 2. Соотношение больных мальчиков
- 8. ПРИМЕРЫ БОЛЕЗНЕЙ Синдром Марфана Синдром Робинова Сирдром Вилльямса Полидактилия Синдактилия Гипертрихоз Ахондроплазия и др.
- 9. Синдром Марфана Впервые описан в 1896 г. Клинические признаки: Наследственное заболевание соединительной ткани , проявляющееся изменениями
- 10. Синдром Марфана Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но
- 11. Синдром Вилльямса Впервые описан в 1961 г. Клинические признаки: Необычное лицо, низкий рост, короткий нос, полные
- 12. Синдром Робинова Впервые описан в 1969 г. Клинические признаки: необычное строение лица, умеренная карликовость, гипоплазия половых
- 13. Полидактилия Клинические признаки: существует два варианта: тип А, при котором дополнительный палец функционален, и тип В,
- 14. Синдактилия Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп. На кистях чаще всего
- 15. Гипертрихоз (люди-волки) Клинические признаки: чрезмерный рост волос на всех частях тела, кроме ладоней и подошв. Других
- 16. Ахондроплазия Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей, большой череп, кисти широкие
- 17. Генные заболевания Аутосомно-рецессивный тип наследования 1. Мутантный ген проявляется только в гомозиготном состоянии. 2.Больной ребенок рождается
- 18. ПРИМЕРЫ БОЛЕЗНЕЙ Фенилкетонурия Микроцефалия Ихтиоз Прогерия Расщелина губы, неба Мукополисахаридоз Альбинизм
- 19. Прогерия Описана в 1886 г. Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения в 8-10 раз.
- 20. Прогерия Я начал стареть, жизнь и так коротка. У многих людей она, как река – Несется
- 21. Расщелина губы, неба Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, деформации первых пальцев кистей, искривление носовой
- 22. Ихтиоз Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по типу гиперкератоза, проявляется
- 23. Мукополисахаридоз Синдром Моркио описан в 1929 г. Клинические признаки: отставание в росте, деформация позвоночника и грудины,
- 24. Фенилкетонурия Клинически впервые описана в 1886 г. Клинические признаки– наличие в моче фенилпировиноградной кислоты и слабоумие.
- 25. Генные заболевания Наследование сцепленное с полом Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ 1. Болеют только мальчики по линии матери.
- 26. ПРИМЕРЫ БОЛЕЗНЕЙ Гемофилия Синдром Элерса-Данло Гидроцефалия Дальтонизм
- 27. Гемофилия Клинические признаки:под- и внутри кожные кровотечения, кровоизлияния в крупные суставы, подкожные и межмышечные гематомы, гематурия,
- 28. Синдром Элерса-Данло Описан в 1657 г. Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза коллагена); кожа тонкая
- 29. Гидроцефалия Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и расхождение костей черепа, диспропорция мозговой
- 30. Дальтонизм Клинические признаки: неспособность различать главным образом красный и зеленый цвета ген дальтонизма передается как рецессивный,
- 31. Хромосомные болезни 1. Хромосомные заболевания связаны с аномалиями числа или структуры хромосом. 2. При каждом заболевании
- 32. ПРИМЕРЫ БОЛЕЗНЕЙ Синдром Шэрешевского-Тернера, Синдром Дауна Синдром «кошачьего крика»
- 33. Синдром Шэрешевского-Тернера Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые вторичные половые признаки, крыловидные кожные складки
- 34. Синдром Дауна Описан в 1866 г. Клинические признаки: умственная отсталость, плоское лицо, монголоид -ный разрез глаз,
- 35. Синдром «кошачьего крика» Описан в 1963 г. Клинические признаки: необычный плач, напоминающий кошачье мяуканье, микроцефалия, антимонголоидный
- 36. Информация к размышлению В 1992 году их число выросло до пяти тысяч В 1986 году было
- 37. Причины изменений в наследственном аппарате человека Спонтанные ошибки при мейозе и репликации ДНК Мутагенные факторы окружающей
- 38. Что надо делать? Профилактика Медико-генетическое консультирование при беременности в возрасте 35 лет и старше, наличии наследственных
- 40. Скачать презентацию

Причины мутаций
Физические факторы (различные виды ионизирующей радиации, ультрафиолетовое излучение)
Химические факторы (инсектициды,
Причины мутаций
Физические факторы (различные виды ионизирующей радиации, ультрафиолетовое излучение)
Химические факторы (инсектициды,
Наследственные заболевания
заболевания человека, обусловленные хромосомными и генными мутациями, передаваемыми по
заболевания человека, обусловленные хромосомными и генными мутациями, передаваемыми по

Историческая справка
В 1929 г. советский генетик, невропатолог С.Н.Давиденко организовал первую
Историческая справка
В 1929 г. советский генетик, невропатолог С.Н.Давиденко организовал первую
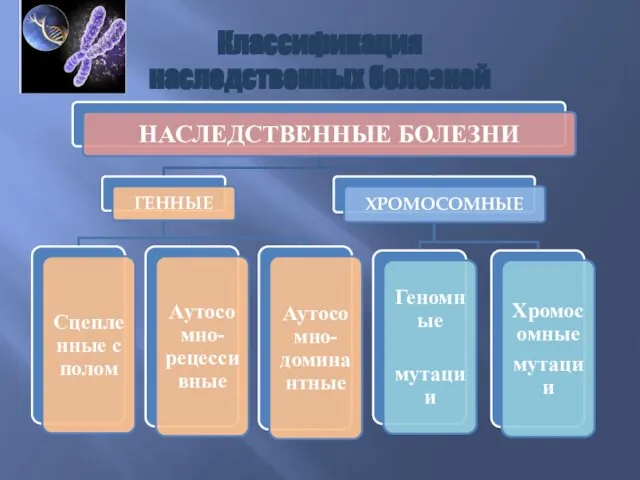
Классификация
наследственных болезней
Классификация
наследственных болезней

Таблица для заполнения Д\з
Таблица для заполнения Д\з

Генные заболевания
Аутосомно-доминирующий
тип наследования
1. Болезнь встечается в каждом
Генные заболевания
Аутосомно-доминирующий
тип наследования
1. Болезнь встечается в каждом

ПРИМЕРЫ БОЛЕЗНЕЙ
Синдром Марфана
Синдром Робинова
Сирдром Вилльямса
Полидактилия
Синдактилия
Гипертрихоз
ПРИМЕРЫ БОЛЕЗНЕЙ
Синдром Марфана
Синдром Робинова
Сирдром Вилльямса
Полидактилия
Синдактилия
Гипертрихоз
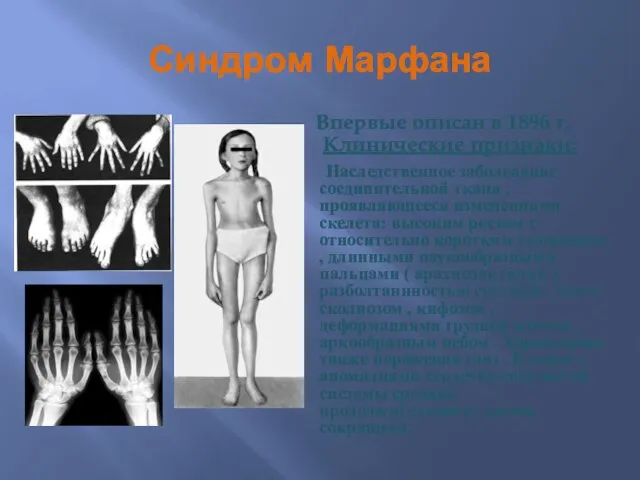
Синдром Марфана
Впервые описан в 1896 г.
Клинические признаки:
Наследственное
Синдром Марфана
Впервые описан в 1896 г.
Клинические признаки:
Наследственное

Синдром Марфана
Высокий выброс адреналина , характерный для заболевания, способствует
Синдром Марфана
Высокий выброс адреналина , характерный для заболевания, способствует

Синдром Вилльямса
Впервые описан в 1961 г.
Клинические признаки:
Необычное лицо, низкий рост, короткий
Синдром Вилльямса
Впервые описан в 1961 г.
Клинические признаки:
Необычное лицо, низкий рост, короткий

Синдром Робинова
Впервые описан в 1969 г.
Клинические признаки: необычное строение лица, умеренная
Синдром Робинова
Впервые описан в 1969 г.
Клинические признаки: необычное строение лица, умеренная

Полидактилия
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
Полидактилия
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
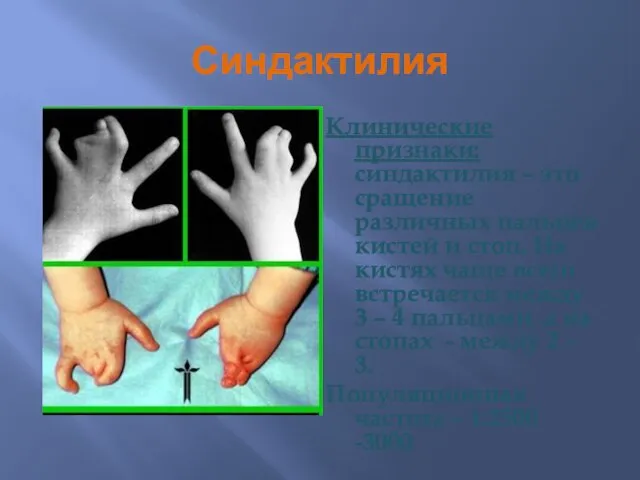
Синдактилия
Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп.
Синдактилия
Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп.

Гипертрихоз
(люди-волки)
Клинические признаки: чрезмерный рост волос на всех частях тела, кроме ладоней
Гипертрихоз
(люди-волки)
Клинические признаки: чрезмерный рост волос на всех частях тела, кроме ладоней

Ахондроплазия
Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей,
Ахондроплазия
Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей,

Генные заболевания
Аутосомно-рецессивный
тип наследования
1. Мутантный ген проявляется только в
Генные заболевания
Аутосомно-рецессивный
тип наследования
1. Мутантный ген проявляется только в

ПРИМЕРЫ БОЛЕЗНЕЙ
Фенилкетонурия
Микроцефалия
Ихтиоз
Прогерия
Расщелина губы, неба
Мукополисахаридоз
Альбинизм
ПРИМЕРЫ БОЛЕЗНЕЙ
Фенилкетонурия
Микроцефалия
Ихтиоз
Прогерия
Расщелина губы, неба
Мукополисахаридоз
Альбинизм
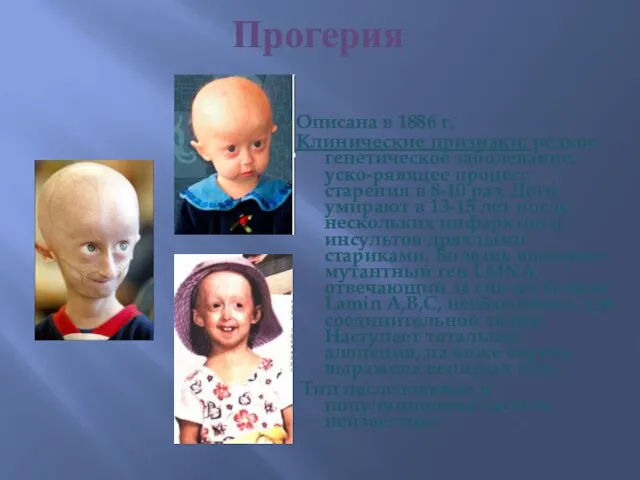
Прогерия
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения
Прогерия
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения

Прогерия
Я начал стареть, жизнь и так коротка.
У многих людей она,
Я начал стареть, жизнь и так коротка. У многих людей она,
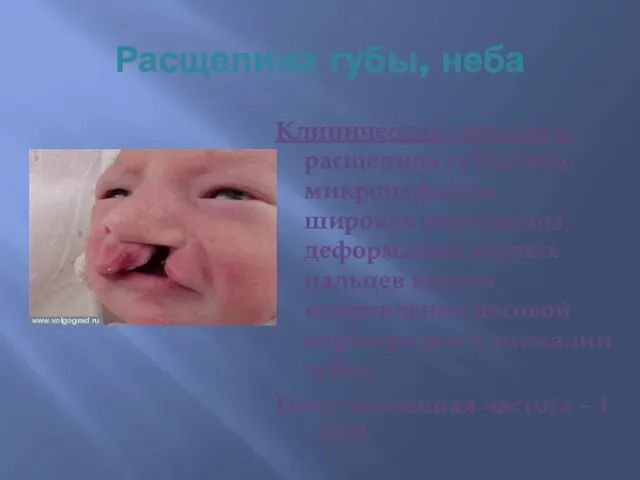
Расщелина губы, неба
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, деформации первых
Расщелина губы, неба
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, деформации первых
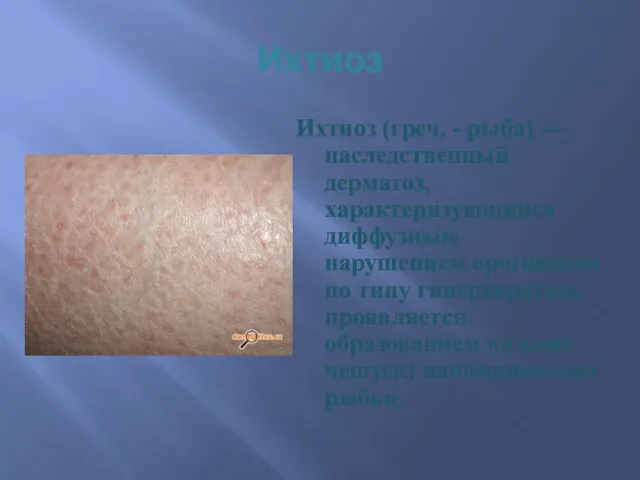
Ихтиоз
Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по
Ихтиоз
Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по
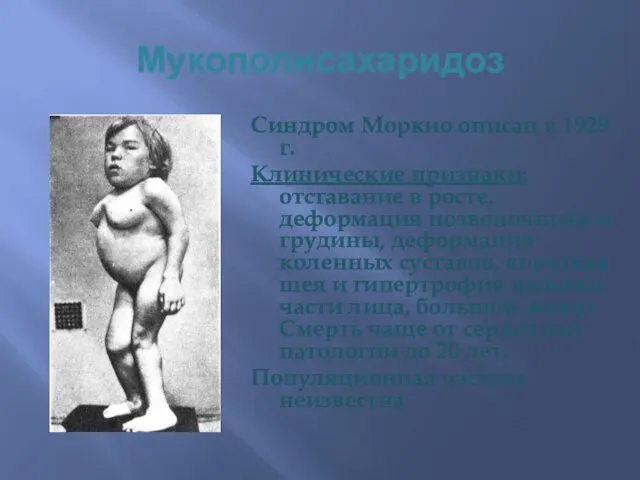
Мукополисахаридоз
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация
Мукополисахаридоз
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация

Фенилкетонурия
Клинически впервые описана в 1886 г.
Клинические признаки– наличие в моче фенилпировиноградной
Фенилкетонурия
Клинически впервые описана в 1886 г.
Клинические признаки– наличие в моче фенилпировиноградной

Генные заболевания
Наследование сцепленное с полом
Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ
1.
Генные заболевания
Наследование сцепленное с полом
Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ
1.

ПРИМЕРЫ БОЛЕЗНЕЙ
Гемофилия
Синдром Элерса-Данло
Гидроцефалия
Дальтонизм
ПРИМЕРЫ БОЛЕЗНЕЙ
Гемофилия
Синдром Элерса-Данло
Гидроцефалия
Дальтонизм
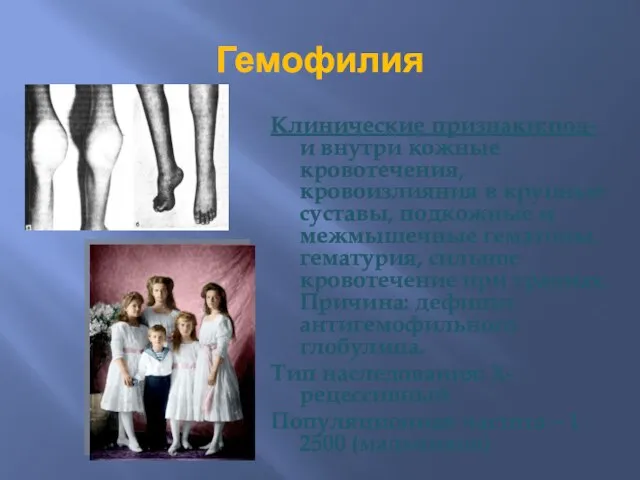
Гемофилия
Клинические признаки:под- и внутри кожные кровотечения, кровоизлияния в крупные суставы, подкожные
Гемофилия
Клинические признаки:под- и внутри кожные кровотечения, кровоизлияния в крупные суставы, подкожные

Синдром Элерса-Данло
Описан в 1657 г.
Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза
Синдром Элерса-Данло
Описан в 1657 г.
Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза

Гидроцефалия
Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и
Гидроцефалия
Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и

Дальтонизм
Клинические признаки: неспособность различать главным образом красный и зеленый цвета
ген
Дальтонизм
Клинические признаки: неспособность различать главным образом красный и зеленый цвета
ген

Хромосомные болезни
1. Хромосомные заболевания связаны с аномалиями числа или структуры
Хромосомные болезни
1. Хромосомные заболевания связаны с аномалиями числа или структуры

ПРИМЕРЫ БОЛЕЗНЕЙ
Синдром Шэрешевского-Тернера,
Синдром Дауна
Синдром «кошачьего крика»
ПРИМЕРЫ БОЛЕЗНЕЙ
Синдром Шэрешевского-Тернера,
Синдром Дауна
Синдром «кошачьего крика»
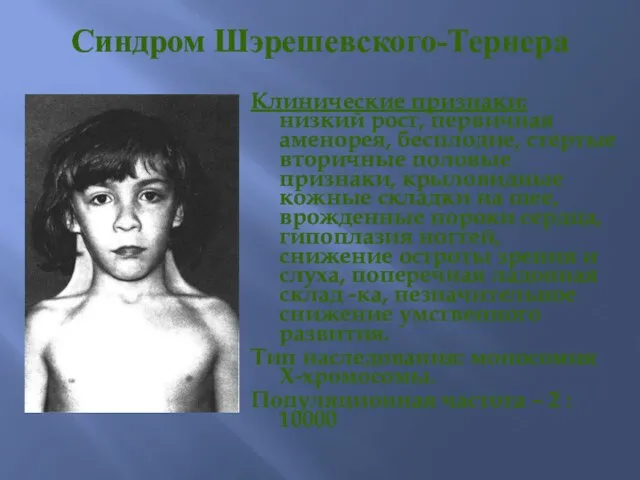
Синдром Шэрешевского-Тернера
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые вторичные половые
Синдром Шэрешевского-Тернера
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые вторичные половые

Синдром Дауна
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское лицо,
Синдром Дауна
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское лицо,
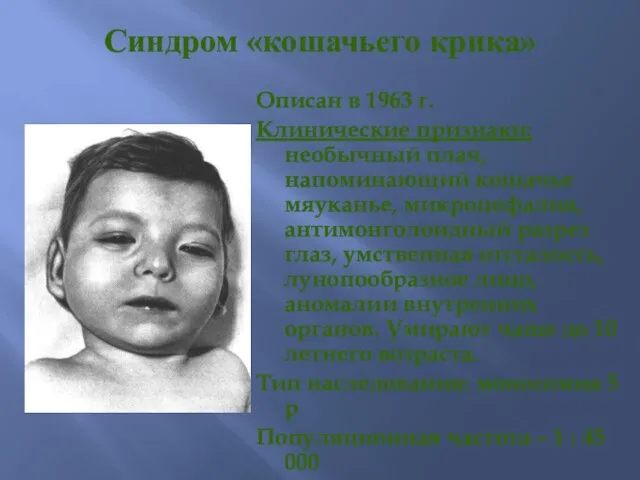
Синдром «кошачьего крика»
Описан в 1963 г.
Клинические признаки: необычный плач, напоминающий кошачье
Синдром «кошачьего крика»
Описан в 1963 г.
Клинические признаки: необычный плач, напоминающий кошачье

Информация к размышлению
В 1992 году их число
выросло до пяти тысяч
В 1986
Информация к размышлению
В 1992 году их число
выросло до пяти тысяч
В 1986
Причины изменений в наследственном аппарате человека
Спонтанные ошибки при мейозе и репликации
Причины изменений в наследственном аппарате человека
Спонтанные ошибки при мейозе и репликации

Что надо делать?
Профилактика
Медико-генетическое консультирование при беременности в возрасте 35 лет
Что надо делать?
Профилактика
Медико-генетическое консультирование при беременности в возрасте 35 лет
 Трофобластична хвороба
Трофобластична хвороба Созылмалы бүйрек жетіспеушілігі, жіктемесі, клиникалық көрінісі
Созылмалы бүйрек жетіспеушілігі, жіктемесі, клиникалық көрінісі Лекарственные растения и фитотерапия
Лекарственные растения и фитотерапия IV Международный студенческий турнир медиков. Задача №3. Статистическая стройность. Теоретический блок. Команда VivatStomat
IV Международный студенческий турнир медиков. Задача №3. Статистическая стройность. Теоретический блок. Команда VivatStomat Шкалы для объективной оценки двигательных функций, а также неврологического статуса у детей со спинальной мышечной атрофией
Шкалы для объективной оценки двигательных функций, а также неврологического статуса у детей со спинальной мышечной атрофией Нәрестенің гемолитикалық ауруы. Басқа сарғаюлармен,анемиялармен,геморрагиялық синдромдармен ажырату
Нәрестенің гемолитикалық ауруы. Басқа сарғаюлармен,анемиялармен,геморрагиялық синдромдармен ажырату Семиотика и синдромология болезней кишечника
Семиотика и синдромология болезней кишечника Что нужно сделать, чтобы ребенок понял: кем быть?
Что нужно сделать, чтобы ребенок понял: кем быть? Артериальды гипертензия. Эналаприл
Артериальды гипертензия. Эналаприл Кафедра пропедевтики внутренних болезней с курсом лучевой диагностики
Кафедра пропедевтики внутренних болезней с курсом лучевой диагностики Психопатологическая семиотика. Практическое занятие №3
Психопатологическая семиотика. Практическое занятие №3 Патофизиология лейкоцитарной системы
Патофизиология лейкоцитарной системы Использование здоровьесберегающих технологий в образовательном процессе
Использование здоровьесберегающих технологий в образовательном процессе Мүмкіндіктері шектеулі науқастармен қарым-қатынас
Мүмкіндіктері шектеулі науқастармен қарым-қатынас Аллергия и в чем она проявляется
Аллергия и в чем она проявляется Поздний гестоз
Поздний гестоз Плодоразрушающие операции. Вакуум-экстракция
Плодоразрушающие операции. Вакуум-экстракция Назначение целевого и результативного компонентов системы психолого-педагогического сопровождения
Назначение целевого и результативного компонентов системы психолого-педагогического сопровождения Психологические аспекты работы с пациентами при отказе от курения
Психологические аспекты работы с пациентами при отказе от курения Терінің маңызы
Терінің маңызы Принципы профилактики коронавирусной инфекции COVID - 19
Принципы профилактики коронавирусной инфекции COVID - 19 Общие основы массажа
Общие основы массажа Агресія дітей. Причини і запобігання
Агресія дітей. Причини і запобігання Внушение народу определенных чувств средствами искусства
Внушение народу определенных чувств средствами искусства Патофизиология опухолевого роста (механизмы канцерогенеза)
Патофизиология опухолевого роста (механизмы канцерогенеза) Скарлатина
Скарлатина Зубы верхней челюсти и техника их моделирования
Зубы верхней челюсти и техника их моделирования Несъемные механически действующие лингвальные брекеты
Несъемные механически действующие лингвальные брекеты