- Свойства электронной волновой функции

Содержание
- 2. молекулярная динамика и метод Монте-Карло неэмпирическая квантовая химия полуэмпирическая квантовая химия квантовая статистическая механика молекулярная механика
- 3. Антисимметричность многоэлектронной волновой функции Электроны обладают собственным моментом количества движения (спином), который в единицах ħ равен
- 4. Ни одна из них не является антисимметричной. Однако связанная с ними функция антисимметрична ( – нормировочный
- 5. Принцип Паули диктует, что две спин-орбитали с одинаковыми пространственными частями (т.е. с одинаковыми квантовыми числами n,
- 6. Заполнение электронами состояний любого атома происходит в порядке возрастания их энергий согласно принципа Паули и каждое
- 7. Детерминант Слейтера Электроны неразличимы, и их перестановка не меняет свойства системы; для волновой функции в виде
- 8. Метод Хартри-Фока Аппроксимация многоэлектронной волновой функции единственным детерминантом Слейтера и использование при ее нахождении ССП метод
- 9. Из условия минимума энергии возникает набор независимых уравнений для каждой одноэлектронной орбитали – уравнений Хартри-Фока: Энергия
- 10. Одноэлектронный интеграл описывает энергию электрона на орбитали в поле ядра без остальных электронов. Двухэлектронный кулоновский интеграл
- 11. Ограниченный и неограниченный методы Хартри-Фока Многоэлектронная волновая функция N-электронной системы должна быть антисимметричной и отвечать определенным
- 12. Собственные значения квадрата спинового момента электронов для разных спиновых состояний Пример - молекулы CH3 в дублетного
- 13. Метод Кона-Шэма Интегрирование проводится по пространственным координатам всех электронов, кроме одного, и по спинам всех электронов;
- 14. Минимизация функционала энергии относительно одноэлектронных функций χi(r) при дополнительном условии их ортонормировки и постоянства числа электронов
- 15. Квантово-химическая трактовка решений одноэлектронных уравнений Физический смысл ХФ энергии орбиталей εi имеют: если удалить с орбитали
- 16. Зависимость потенциалов ионизации элементов от атомного номера В пределах периода с увеличением атомного номера потенциалы ионизации
- 17. Приравнивая нулю дифференциал функционала электронной энергии d{E[ρ] – μN[ρ(r)]}Vяд = const = 0, (N = ∫
- 18. Зависимость энергии атома от числа электронов μ характеризует наклон кривой E = E(N). Производная в конечно-разностном
- 19. Определенная таким образом величина называется абсолютной электроотрицательностью (Parr, R. G. at all. 1978). Пирсон (1983), действуя
- 20. Можно также определить индекс электрофильности (Parr R. G. at all. 1999) = = , величина которого
- 21. Зависимости жесткости η, индекса электрофильности ω и поляризуемости от порядкового номера элемента
- 22. Атомы с закрытыми оболочками или подоболочками имеют большую жесткость и малую поляризуемость . В каждом периоде
- 24. Скачать презентацию
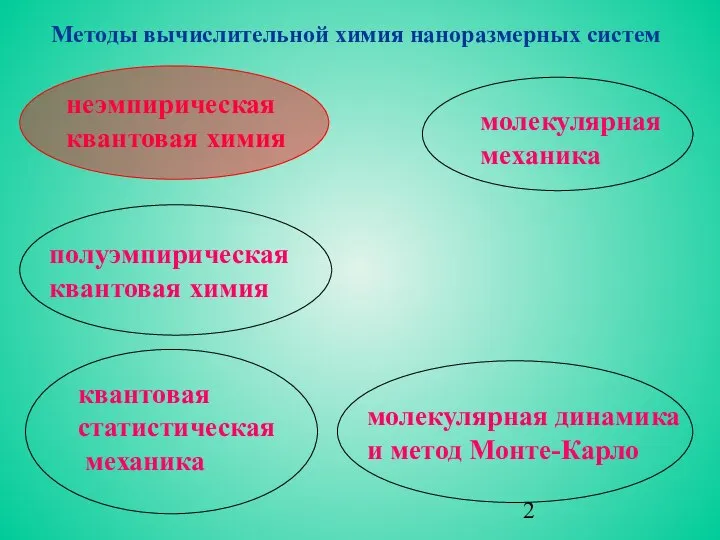
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия

Антисимметричность многоэлектронной волновой функции
Электроны обладают собственным моментом количества движения (спином), который
Антисимметричность многоэлектронной волновой функции
Электроны обладают собственным моментом количества движения (спином), который

Ни одна из них не является антисимметричной. Однако связанная с ними
Ни одна из них не является антисимметричной. Однако связанная с ними

Принцип Паули диктует, что две спин-орбитали с одинаковыми пространственными частями (т.е.
Принцип Паули диктует, что две спин-орбитали с одинаковыми пространственными частями (т.е.

Заполнение электронами состояний любого атома происходит в порядке возрастания их энергий
Заполнение электронами состояний любого атома происходит в порядке возрастания их энергий

Детерминант Слейтера
Электроны неразличимы, и их перестановка не меняет свойства системы;
Детерминант Слейтера
Электроны неразличимы, и их перестановка не меняет свойства системы;

Метод Хартри-Фока
Аппроксимация многоэлектронной волновой функции единственным детерминантом Слейтера и использование при
Метод Хартри-Фока
Аппроксимация многоэлектронной волновой функции единственным детерминантом Слейтера и использование при

Из условия минимума энергии возникает набор независимых уравнений для каждой одноэлектронной
Из условия минимума энергии возникает набор независимых уравнений для каждой одноэлектронной

Одноэлектронный интеграл описывает энергию электрона на орбитали в поле ядра без
Одноэлектронный интеграл описывает энергию электрона на орбитали в поле ядра без

Ограниченный и неограниченный методы Хартри-Фока
Многоэлектронная волновая функция N-электронной системы должна
Ограниченный и неограниченный методы Хартри-Фока
Многоэлектронная волновая функция N-электронной системы должна
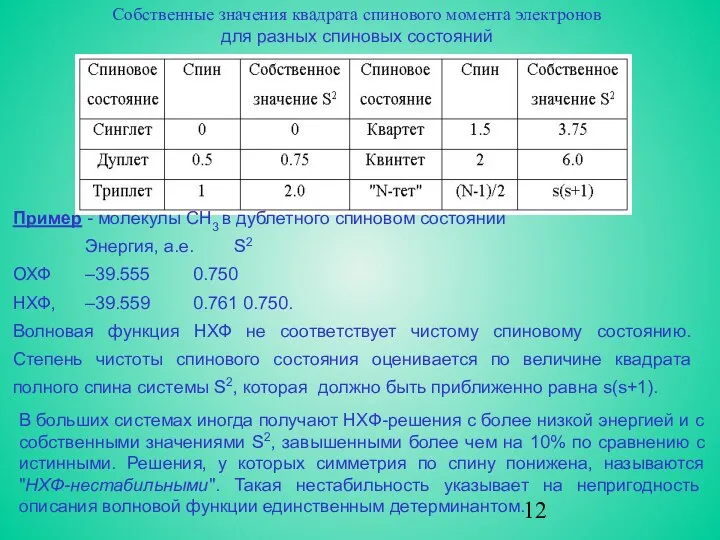
Собственные значения квадрата спинового момента электронов
для разных спиновых состояний
Пример -
Собственные значения квадрата спинового момента электронов
для разных спиновых состояний
Пример -

Метод Кона-Шэма
Интегрирование проводится по пространственным координатам всех электронов, кроме одного,
Метод Кона-Шэма
Интегрирование проводится по пространственным координатам всех электронов, кроме одного,

Минимизация функционала энергии относительно одноэлектронных функций χi(r) при дополнительном условии их
Минимизация функционала энергии относительно одноэлектронных функций χi(r) при дополнительном условии их

Квантово-химическая трактовка решений
одноэлектронных уравнений
Физический смысл ХФ энергии орбиталей εi имеют: если
Квантово-химическая трактовка решений
одноэлектронных уравнений
Физический смысл ХФ энергии орбиталей εi имеют: если
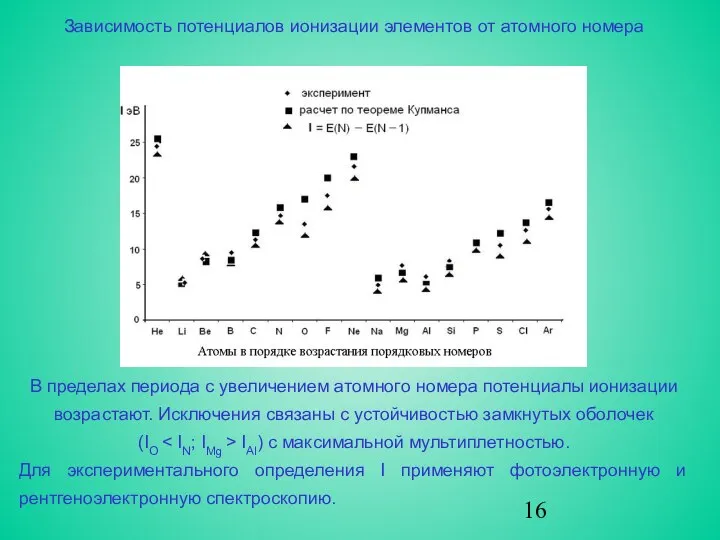
Зависимость потенциалов ионизации элементов от атомного номера
В пределах периода с
Зависимость потенциалов ионизации элементов от атомного номера
В пределах периода с
![Приравнивая нулю дифференциал функционала электронной энергии d{E[ρ] – μN[ρ(r)]}Vяд = const](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1297931/slide-16.jpg)
Приравнивая нулю дифференциал функционала электронной энергии d{E[ρ] – μN[ρ(r)]}Vяд = const
Приравнивая нулю дифференциал функционала электронной энергии d{E[ρ] – μN[ρ(r)]}Vяд = const
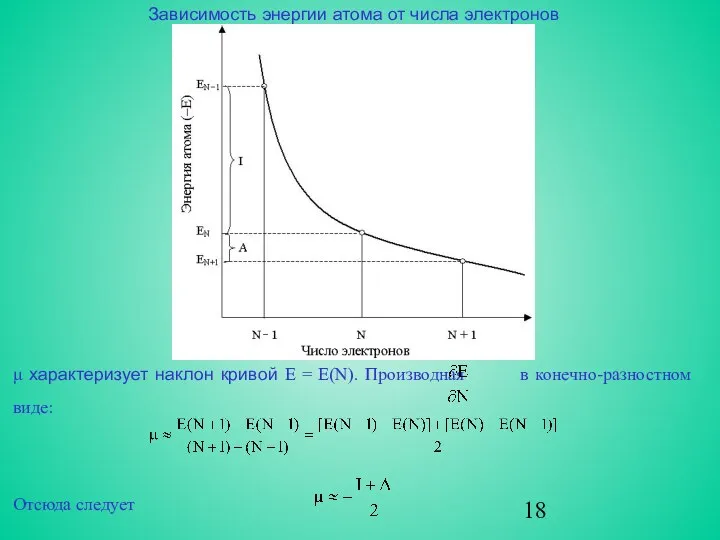
Зависимость энергии атома от числа электронов
μ характеризует наклон кривой E =
Зависимость энергии атома от числа электронов
μ характеризует наклон кривой E =

Определенная таким образом величина
называется абсолютной электроотрицательностью (Parr, R. G. at all.
Определенная таким образом величина
называется абсолютной электроотрицательностью (Parr, R. G. at all.

Можно также определить индекс электрофильности (Parr R. G. at all. 1999)
Можно также определить индекс электрофильности (Parr R. G. at all. 1999)
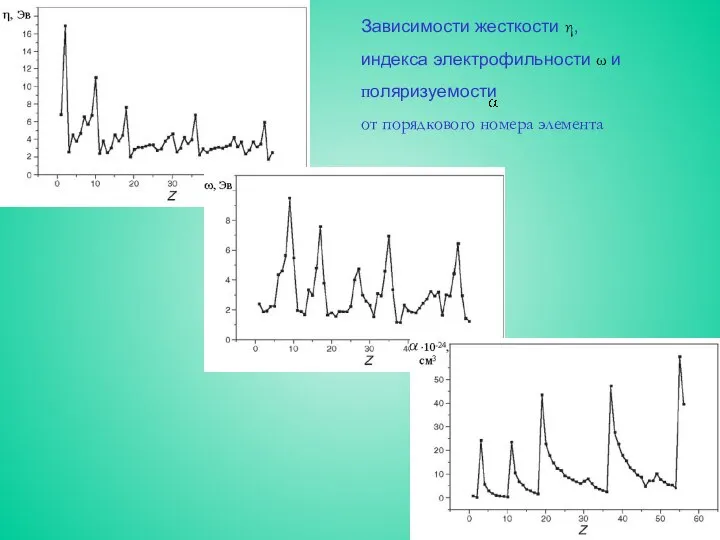
Зависимости жесткости η,
индекса электрофильности ω и
поляризуемости
от порядкового номера элемента
Зависимости жесткости η,
индекса электрофильности ω и
поляризуемости
от порядкового номера элемента

Атомы с закрытыми оболочками или подоболочками имеют большую жесткость и малую
Атомы с закрытыми оболочками или подоболочками имеют большую жесткость и малую
 Основы технологии оклейки стен обоями
Основы технологии оклейки стен обоями По статистике* женщины с маленькой грудью 18 раз в день думают, что большая грудь позволит им найти любимого. - презентация
По статистике* женщины с маленькой грудью 18 раз в день думают, что большая грудь позволит им найти любимого. - презентация Литературная викторина Автор презентации Клюквина В А
Литературная викторина Автор презентации Клюквина В А Свойства некоторых аналоговых фильтров
Свойства некоторых аналоговых фильтров  Types of houses
Types of houses Визуальная система программирования Delphi. Язык программирования Object Pascal
Визуальная система программирования Delphi. Язык программирования Object Pascal СМИ в избирательных процессах
СМИ в избирательных процессах Как выбрать подрядчика по контекстной рекламе: преимущества и недостатки самостоятельного ведения
Как выбрать подрядчика по контекстной рекламе: преимущества и недостатки самостоятельного ведения  Грех и его трактовка в разных религиях
Грех и его трактовка в разных религиях Взаимное положение прямых
Взаимное положение прямых Презентация Жизнь и творчество И.С, Баха
Презентация Жизнь и творчество И.С, Баха А. Т. Аверченко 1881-1925
А. Т. Аверченко 1881-1925 Личное и групповое туристское снаряжение и уход за ним. Индивидуальный и групповой ремонтный набор
Личное и групповое туристское снаряжение и уход за ним. Индивидуальный и групповой ремонтный набор Экскурсия по Афинам
Экскурсия по Афинам INTEL - INTegrated circuits and ELectronics
INTEL - INTegrated circuits and ELectronics Расчет системы охлаждения
Расчет системы охлаждения  Периодизация развития художественной культуры. Стили эпохи
Периодизация развития художественной культуры. Стили эпохи Беседа по картине В.М. Васнецова «Богатыри» Автор: Сосновцева Валентина Николаевна – учитель начальных классов МОУ «СОШ с. Елшанк
Беседа по картине В.М. Васнецова «Богатыри» Автор: Сосновцева Валентина Николаевна – учитель начальных классов МОУ «СОШ с. Елшанк «Роль характера взаимоотношений между родителями и детьми в развитии личности ребенка»
«Роль характера взаимоотношений между родителями и детьми в развитии личности ребенка» Разработка сетевых приложений
Разработка сетевых приложений Исследование развития скоростных способностей у школьников 13-14 лет на уроках баскетбола
Исследование развития скоростных способностей у школьников 13-14 лет на уроках баскетбола Построение топографического плана по результатам нивелирования по квадратам
Построение топографического плана по результатам нивелирования по квадратам УЧЕТ РАСХОДОВ ПО СТРОИТЕЛЬНЫМ МАШИНАМ И МЕХАНИЗМАМ
УЧЕТ РАСХОДОВ ПО СТРОИТЕЛЬНЫМ МАШИНАМ И МЕХАНИЗМАМ Личность педагога в современной школе Личность воспитателя значит все в деле воспитания К.Д Уш
Личность педагога в современной школе Личность воспитателя значит все в деле воспитания К.Д Уш Презентация "1 ВВЕДЕНИЕ В МАКРОЭКОНОМИКУ" - скачать презентации по Экономике
Презентация "1 ВВЕДЕНИЕ В МАКРОЭКОНОМИКУ" - скачать презентации по Экономике Семейное увлечение хоккей
Семейное увлечение хоккей Traditions and holidays of Great Britain
Traditions and holidays of Great Britain Полезные ископаемые Презентацию подготовила Сковородина Елена Павловна, учитель начальны
Полезные ископаемые Презентацию подготовила Сковородина Елена Павловна, учитель начальны