- Формальная кинетика. Предмет химической кинетики

Содержание
- 2. Такое кажущееся противоречие между теоретическими предсказаниями и практическими результатами обусловлено тем, что в термодинамике учитывается только
- 3. Можно выделить две главные задачи химической кинетики, определяющие ее практическое и теоретическое значение: экспериментальное исследование скорости
- 8. В связи с этим для элементарных реакций вводится понятие молекулярность – число молекул, принимающих участие в
- 11. Простые необратимые реакции В системе одновременно и независимо могут протекать реакции с различной скоростью, но в
- 14. Рис. 1. Зависимость концентраций прореагировавшего и оставшегося вещества от времени
- 26. Методы определения порядка реакции При определении порядка реакции вначале находят порядок по каждому из реагирующих веществ.
- 27. Метод графического подбора. Как следует из уравнения (10) для реакции первого порядка выполняется линейная зависимость в
- 28. 2. Метод аналитического подбора уравнения заключается в том, что проводится расчет константы скорости путем подстановки экспериментальных
- 29. Рис. 3. Определение порядка по периоду полуреакции
- 30. 4. Графический метод определения порядка. Скорость реакции n-ого порядка по данному веществу равна v = kcn
- 31. Рис. 4. Определение порядка реакции графическим методом: a) нахождение скорости; б) нахождение порядка и константы скорости
- 44. Рис. 5. Зависимость концентрации веществ от времени в последовательных реакциях
- 48. Метод стационарных концентраций В рассмотренном выше простейшем случае двух последовательных реакций первого порядка получены уравнения для
- 53. В заключение заметим, что метод стационарных концентраций не является совершенно строгим, его применение ограничивается выполнением условий
- 60. Как видно из уравнения (97), логарифм константы скорости является линейной функцией обратной температуры. Поэтому для экспериментального
- 61. Рис. 6. Зависимость lnk от 1/T
- 63. Рис. 7. Зависимость lnk от 1/T для параллельных реакций
- 69. Величина ED небольшая (5 ÷ 10 кДж/моль), т.е. ED Так как энергия активации диффузии невелика, то
- 71. Скачать презентацию

Такое кажущееся противоречие между теоретическими предсказаниями и практическими результатами обусловлено тем,
Такое кажущееся противоречие между теоретическими предсказаниями и практическими результатами обусловлено тем,

Можно выделить две главные задачи химической кинетики, определяющие ее практическое и
Можно выделить две главные задачи химической кинетики, определяющие ее практическое и





В связи с этим для элементарных реакций вводится понятие молекулярность –
В связи с этим для элементарных реакций вводится понятие молекулярность –



Простые необратимые реакции
В системе одновременно и независимо могут протекать реакции с
Простые необратимые реакции
В системе одновременно и независимо могут протекать реакции с


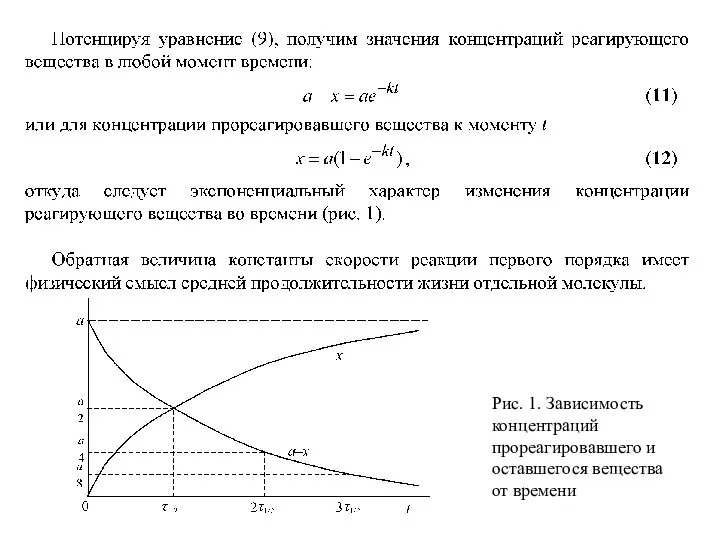
Рис. 1. Зависимость концентраций
прореагировавшего и оставшегося вещества
от времени
Рис. 1. Зависимость концентраций
прореагировавшего и оставшегося вещества
от времени












Методы определения порядка реакции
При определении порядка реакции вначале находят порядок по
Методы определения порядка реакции
При определении порядка реакции вначале находят порядок по

Метод графического подбора.
Как следует из уравнения (10) для реакции первого
Метод графического подбора.
Как следует из уравнения (10) для реакции первого

2. Метод аналитического подбора уравнения заключается в том, что проводится расчет
2. Метод аналитического подбора уравнения заключается в том, что проводится расчет

Рис. 3. Определение порядка
по периоду полуреакции
Рис. 3. Определение порядка
по периоду полуреакции

4. Графический метод определения порядка.
Скорость реакции n-ого порядка по данному
4. Графический метод определения порядка.
Скорость реакции n-ого порядка по данному
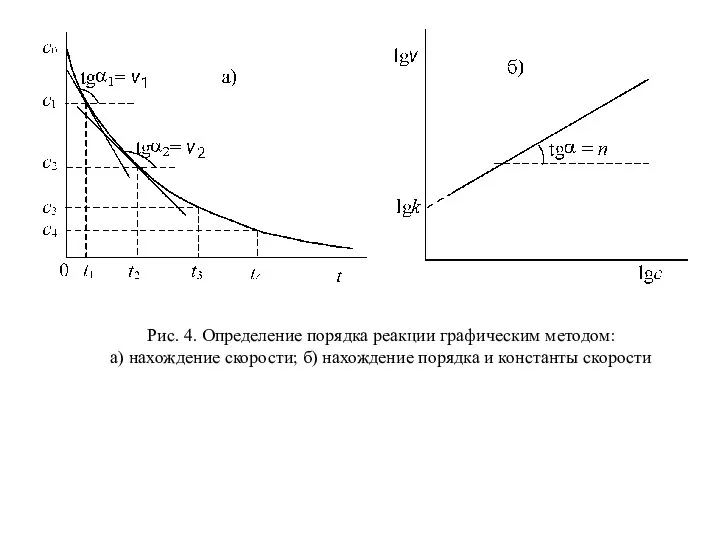
Рис. 4. Определение порядка реакции графическим методом:
a) нахождение скорости; б) нахождение
Рис. 4. Определение порядка реакции графическим методом:
a) нахождение скорости; б) нахождение












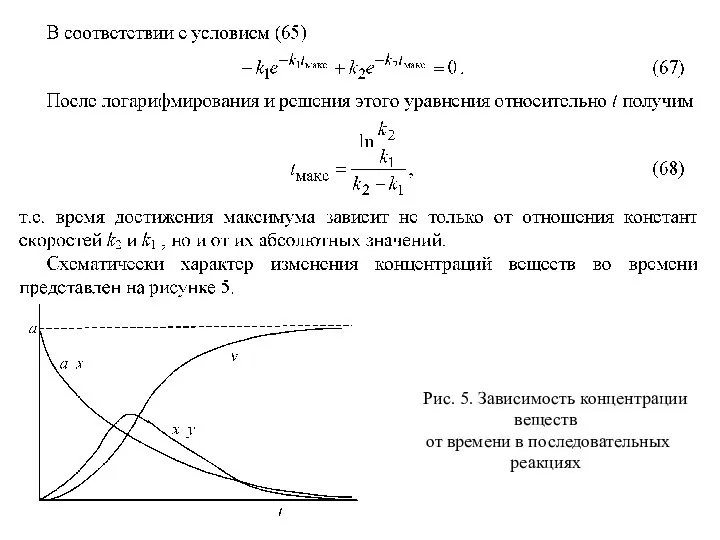
Рис. 5. Зависимость концентрации веществ
от времени в последовательных реакциях
Рис. 5. Зависимость концентрации веществ
от времени в последовательных реакциях




Метод стационарных концентраций
В рассмотренном выше простейшем случае двух последовательных реакций первого
Метод стационарных концентраций
В рассмотренном выше простейшем случае двух последовательных реакций первого





В заключение заметим, что метод стационарных концентраций не является совершенно строгим,
В заключение заметим, что метод стационарных концентраций не является совершенно строгим,







Как видно из уравнения (97), логарифм константы скорости является линейной функцией
Как видно из уравнения (97), логарифм константы скорости является линейной функцией
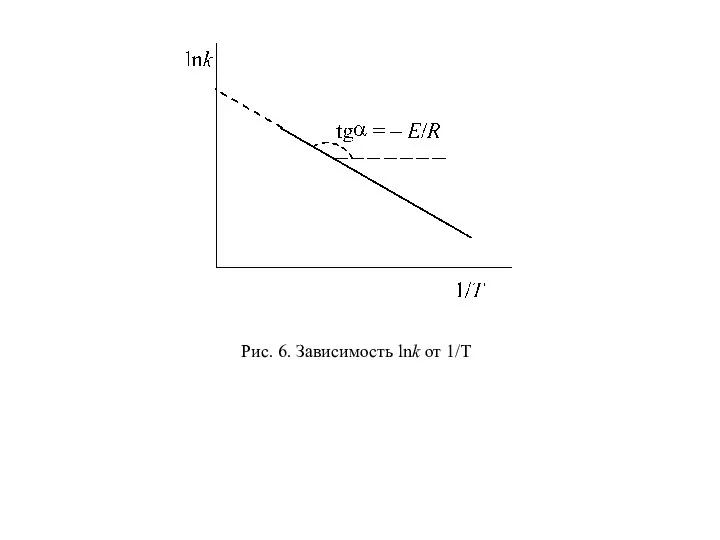
Рис. 6. Зависимость lnk от 1/T
Рис. 6. Зависимость lnk от 1/T

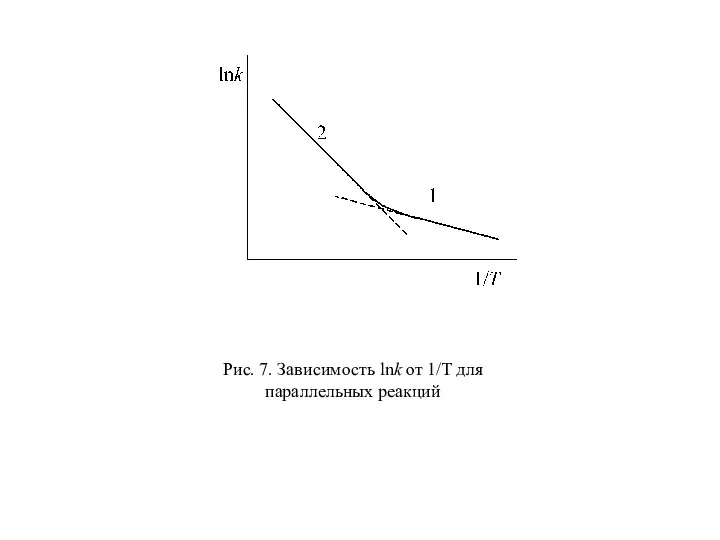
Рис. 7. Зависимость lnk от 1/T для параллельных реакций
Рис. 7. Зависимость lnk от 1/T для параллельных реакций






Величина ED небольшая (5 ÷ 10 кДж/моль), т.е. ED << EA
Величина ED небольшая (5 ÷ 10 кДж/моль), т.е. ED << EA
 Физико-химия поверхностных явлений. Основы адсорбционной терапии
Физико-химия поверхностных явлений. Основы адсорбционной терапии Класифікація неорганічних сполук
Класифікація неорганічних сполук Уроки зельеварения. Задача 6
Уроки зельеварения. Задача 6 Предмет, задачи, понятия, законы химии
Предмет, задачи, понятия, законы химии Термодинамические свойства газов
Термодинамические свойства газов Химическое равновесие. Необратимые и обратимые реакции
Химическое равновесие. Необратимые и обратимые реакции Морфология тел полезных ископаемых
Морфология тел полезных ископаемых Презентация по Химии "Белки" - скачать смотреть бесплатно_
Презентация по Химии "Белки" - скачать смотреть бесплатно_ Применение аммиака и солей аммония
Применение аммиака и солей аммония Кислотно-основные равновесия
Кислотно-основные равновесия Поверхностное упрочнение деталей. (Лекция 11)
Поверхностное упрочнение деталей. (Лекция 11) Презентация по химии на тему: «Применение кислорода»
Презентация по химии на тему: «Применение кислорода» Перманганат калия
Перманганат калия Спирты (карбинолы)
Спирты (карбинолы) Методы исследования наносистем и наноматериалов. Классификация физико-химических методов исследования
Методы исследования наносистем и наноматериалов. Классификация физико-химических методов исследования Отраслевые стандарты качества лекарственных средств: GMP, GLP, GSP, GDP, GPP, GCP и другие
Отраслевые стандарты качества лекарственных средств: GMP, GLP, GSP, GDP, GPP, GCP и другие Обмен веществ. Метаболизм и его функции
Обмен веществ. Метаболизм и его функции Идентификация органических веществ
Идентификация органических веществ Жировая ткань
Жировая ткань Презентация по Химии "Анализ тенденций развития химии" - скачать смотреть
Презентация по Химии "Анализ тенденций развития химии" - скачать смотреть  Ауылшаруашылық дақылдарының тұқымдарын фунгецидтермен улау және химиялық қорғау тәсілдерінің биологиялық
Ауылшаруашылық дақылдарының тұқымдарын фунгецидтермен улау және химиялық қорғау тәсілдерінің биологиялық Кольорові метали
Кольорові метали 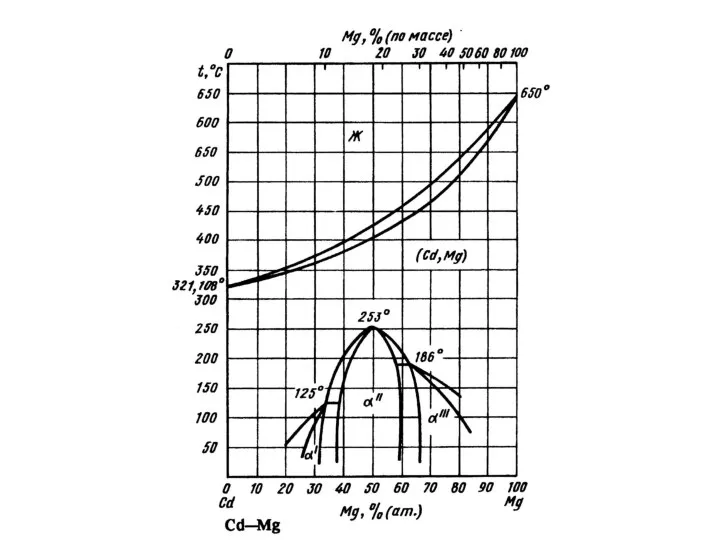 Промежуточные фазы
Промежуточные фазы Смеси и растворы
Смеси и растворы Липидтердің метаболизмі
Липидтердің метаболизмі Аттестационная работа. Образовательная программа элективного курса Химия вокруг нас
Аттестационная работа. Образовательная программа элективного курса Химия вокруг нас В таблиці я ― відомий елемент, Та літеру зміни з одного боку, Переконаєшся в один момент, Що я не елемент вже, а протока. Метаграми
В таблиці я ― відомий елемент, Та літеру зміни з одного боку, Переконаєшся в один момент, Що я не елемент вже, а протока. Метаграми  ЕГЭ по химии
ЕГЭ по химии