- Генные болезни. Генетические и клинические аспекты. (Лекция 6)

Содержание
- 2. ГЕННЫЕ БОЛЕЗНИ – это группа наследственных заболеваний человека, в основе развития которых лежат мутации отдельных, или
- 3. ГЕННЫЕ БОЛЕЗНИ носят врожденный характер манифестируют в любом возрасте проявляются определенной, часто поргрессирующей клинической симптоматикой сопровождаются
- 4. ГЕННЫЕ БОЛЕЗНИ Делятся на две основные группы: Наследственные болезни обмена веществ – НБО Наследственные болезни, проявляющиеся
- 5. ТИПЫ НАСЛЕДОВАНИЯ НБО Аутосомно-рецессивный Большинство НБО Аутосомно-доминантный Острая перемежающаяся порфирия (порфобилиногендезаминаза), семейная гиперхолестеринемия (LDL-рецептор), семейный сфероцитоз
- 6. ПРИМЕРЫ НБО И ИХ ЧАСТОТА в отдельных этнических группах
- 7. БИОХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ НБО Болезни углеводного обмена Болезни обмена аминокислот Болезни обмена органических кислот Болезни обмена жирных
- 8. КЛИНИЧЕСКАЯ КЛАССИФИКАЦИЯ НБО
- 9. ТРУДНОСТИ ПРИ ДИАГНОСТИКЕ НБО Сходство клинической картины с частыми приобретенными состояниями (Энцефалопатия при НБО – арбовирусная,
- 10. ВАРИАНТЫ КЛИНИЧЕСКОЙ МАНИФЕСТАЦИИ НБО Острые тяжелые расстройства в неонатальном периоде Поздно появляющиеся острые или повторяющиеся симптомы
- 11. НБО С ПСИХИАТРИЧЕСКИМИ И ПОВЕДЕНЧЕСКИМИ РАССТРОЙСТВАМИ
- 12. ЭТАПЫ ДИАГНОСТИКИ РЕДКИХ НБО Стандартные скрининговые тесты Количественные исследования Нагрузочные пробы Исследования вовлеченности в патологический процесс
- 13. СПИСОК НБО, ПОДЛЕЖАЩИХ ДНК-ДИАГНОСТИКЕ - Адреногенитальный синдром - Адренолейкодистрофия - Альбинизм - Болезнь Вильсона-Коновалова - Болезнь
- 14. ПАТОМОРФОЛОГОГИЧЕСКИЕ ИССЛЕДОВАНИЯ
- 15. ПРИНЦИПЫ ЛЕЧЕНИЯ НБО Контроль за накоплением субстрата 1.1. Диета с удалением субстрата 1.2. Контроль за эндогенной
- 16. ДИЕТА С УДАЛЕНИЕМ СУБСТРАТА
- 17. УСКОРЕННОЕ ВЫВЕДЕНИЕ СУБСТРАТА Диализ (перитонеальный диализ, гемодиализ, длительная вено-венозная гемофильтрация) Использование бензоата, фенилацетата и фенилбутирата натрия
- 18. КОНТРОЛЬ ЗА ЭНДОГЕННОЙ ПРОДУКЦИЕЙ СУБСТРАТА Болезни аминокислотного обмена и дефекты цикла мочевины во время сопутствующих заболеваний
- 19. ПРИМЕРЫ КОФАКТОРОВ, ИСПОЛЬЗУЕМЫХ ПРИ ЛЕЧЕНИИ НЕКОТОРЫХ НБО
- 20. ЗАМЕСТИТЕЛЬНАЯ ФЕРМЕНТОТЕРАПИЯ Болезнь Гоше – препарат церезим
- 21. ПРИМЕРЫ ТРАСПЛАНТАЦИИ ОРГАНОВ ПРИ НБО Трансплантация печени: Недостаточность α1-антитрипсина, наследственная тирозинемия, некоторые формы болезни Вильсона-Коновалова, гликогенозы
- 22. ПРИНЦИПЫ ИНДИВИДУАЛЬНОЙ, СЕМЕЙНОЙ И МАССОВОЙ ПРОФИЛАКТИКИ НБО 1. Своевременная диагностика, лабораторное подтверждение и лечение нозологической формы
- 23. ПРИМЕРЫ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ ОБМЕНА ВЕЩЕСТВ
- 24. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА ЛАКТАЗНАЯ НЕДОСТАТОЧНОСТЬ Дефектый фермент: лактаза (дисахаридаза) расщепляет молочный сахар-лактозу на глюкозу и галактозу
- 25. Генетически и клинически гетерогенная группа заболеваний Дефектый фермент: галактокиназа - GALK, галактозо-1-фосфат-уридилтрансфераза - GALT, уридиндифосфат-галактозо-4-эпимераза -
- 26. Мутантный ген – GALT на 9р13 Дефектный фермент - галактозо-1-фосфатуридилтрансфераза Тип наследования: АР (аутосомно-рецессивный) Манифестация заболевания:обычно
- 27. Вторичные осложнения: -спленомегалия, -нарушение свертывания крови, -картина геморрагического диатеза и гемолитической анемии, -симптомы почечной недостаточности -
- 28. Критерии диагноза: повышенные концентрации галактозы и/или галактозо-1-фосфата, снижение содержания глюкозы и толерантности к ней, повышение уровня
- 29. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА ГАЛАКТОЗЕМИЯ I типа Г.А., 2001 г.р. Диагноз при рождении: здорова Диагноз клинический: Гепатоспленомегалия.
- 30. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА ГАЛАКТОЗЕМИЯ II типа Мутантный ген – GALК на 17q24 Дефектный фермент - галактокиназа
- 31. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА ГАЛАКТОЗЕМИЯ II типа С.Э., 1998 г.р. Диагноз при рождении: ВУ гипоксия плода Диагноз
- 32. Мутантный ген – ALDA картирован на 9q21.3-22.2 Дефектый фермент - альдолаза В (фруктозо-1-фосфатальдолаза), в норме осуществляет
- 33. Клинические проявления рвота отвращение к пище содержащей фруктозу гепатомегалия, желтуха слабость, вялость гипервозбудимость судороги, обусловленные гипогликемией
- 34. Лабораторные данные: -фруктозурия, -альбуминурия, -гипераминоацидурия, -гиперфруктоземия после нагрузки фруктозой, гипогликемия Лечение: диета не содержащая фруктозу и
- 35. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА ФРУКТОЗЕМИЯ К.М., 1987 г.р. Диагноз при рождении: здоров. Болеет с раннего возраста. Диагноз
- 36. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА ГЛЮКОЦЕРЕБРОЗИДОЗЫ
- 37. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА БОЛЕЗНЬ ГОШЕ АР-наследование Ген GBA, 1q21-q31 Измененный фермент - глюкоцереброзидаза Г.А., 2003 г.р.
- 38. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА МУКОПОЛИСАХАРИДОЗЫ
- 39. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА МУКОПОЛИСАХАРИДОЗ VI типа АР-наследование Ген ARSB, 5q13.3 Измененный фермент - арилсульфатаза В Т.М.,
- 40. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА ГЛИКОГЕНОЗЫ Тип I - болезнь Гирке (10 клинических форм) Тип II - болезнь
- 41. Болезни углеводного обмена Гликогенозы - Тип I (болезнь Гирке) Дефектый фермент: глюкозо-6-фосфотаза, катализирует конечную реакцию глюконеогенеза
- 42. Болезни углеводного обмена Гликогенозы - Тип I (болезнь Гирке) Большой живот, гепатомегалия, нефромегалия гипогликемия, часто с
- 43. Болезни углеводного обмена Гликогенозы - Тип II (болезнь Помпе) Дефектый фермент: α-D-глюкозидаза участвует в гидролизе гликогена
- 44. Болезни углеводного обмена Гликогенозы - Тип II (болезнь Помпе) Мышечная слабость, включая дыхательную мускулатуру снижение двигательной
- 45. Болезни углеводного обмена Гликогенозы - Тип III (болезнь Кори) Дефектый фермент: амило-1,6-глюкозидаза, фермент укорачивающий цепи. Тип
- 46. Болезни углеводного обмена Гликогенозы - Тип III (болезнь Кори) гепатомегалия гипогликемия гиперлипидемия толерантность к глюкозе и
- 47. Болезни углеводного обмена Гликогенозы - Тип IV (болезнь Андерсена) Дефектый фермент: амило-1,4:1,6-глюкотрансфераза, ветвящий фермент участвует в
- 48. Болезни углеводного обмена Гликогенозы - Тип IV (болезнь Андерсена) Рвота диарея нарушение вскармливания задержка физического развития
- 49. Болезни углеводного обмена Гликогенозы - Тип V (болезнь Мак-Ардла) Дефектый фермент: мышечная фосфорилаза Тип наследования: АР
- 50. Болезни углеводного обмена Гликогенозы - Тип VI (болезнь Герса) Дефектный фермент: фосфорилаза Тип наследования: АР Клинические
- 51. Болезни обмена аминокислот Фенилкетонурия I, II, III типы, материнская ФКУ Тирозинемия I, II, III типы Альбинизм
- 56. НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ ФЕНИЛКЕТОНУРИЯ С.А., 1983 г.р.; Диагноз ФКУ в 2,5 года (г.Санкт-петербург); Лечение в течение
- 57. НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ ТИРОЗИНЕМИЯ АР-наследование Ген FAH - 15q23-25 Измененный фермент - фумарилацетоацетаза Б.Я., 1998 г.р.;
- 58. НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ АЛЬБИНИЗМ АР-наследование Ген TYR - 11q14-q21 Измененный фермент – тирозиназа К.О., 1992 г.р.
- 60. Скачать презентацию

ГЕННЫЕ БОЛЕЗНИ –
это группа наследственных заболеваний человека, в основе
ГЕННЫЕ БОЛЕЗНИ –
это группа наследственных заболеваний человека, в основе

ГЕННЫЕ БОЛЕЗНИ
носят врожденный характер
манифестируют в любом возрасте
проявляются определенной, часто поргрессирующей
ГЕННЫЕ БОЛЕЗНИ
носят врожденный характер
манифестируют в любом возрасте
проявляются определенной, часто поргрессирующей

ГЕННЫЕ БОЛЕЗНИ
Делятся на две основные группы:
Наследственные болезни обмена веществ – НБО
Наследственные
ГЕННЫЕ БОЛЕЗНИ
Делятся на две основные группы:
Наследственные болезни обмена веществ – НБО
Наследственные

ТИПЫ НАСЛЕДОВАНИЯ НБО
Аутосомно-рецессивный
Большинство НБО
Аутосомно-доминантный
Острая перемежающаяся порфирия (порфобилиногендезаминаза), семейная
ТИПЫ НАСЛЕДОВАНИЯ НБО
Аутосомно-рецессивный
Большинство НБО
Аутосомно-доминантный
Острая перемежающаяся порфирия (порфобилиногендезаминаза), семейная

ПРИМЕРЫ НБО И ИХ ЧАСТОТА в отдельных этнических группах
ПРИМЕРЫ НБО И ИХ ЧАСТОТА в отдельных этнических группах

БИОХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ НБО
Болезни углеводного обмена
Болезни обмена аминокислот
Болезни обмена органических кислот
Болезни
БИОХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ НБО
Болезни углеводного обмена
Болезни обмена аминокислот
Болезни обмена органических кислот
Болезни

КЛИНИЧЕСКАЯ КЛАССИФИКАЦИЯ НБО
КЛИНИЧЕСКАЯ КЛАССИФИКАЦИЯ НБО

ТРУДНОСТИ ПРИ ДИАГНОСТИКЕ НБО
Сходство клинической картины с частыми приобретенными состояниями
(Энцефалопатия
ТРУДНОСТИ ПРИ ДИАГНОСТИКЕ НБО
Сходство клинической картины с частыми приобретенными состояниями
(Энцефалопатия

ВАРИАНТЫ КЛИНИЧЕСКОЙ МАНИФЕСТАЦИИ НБО
Острые тяжелые расстройства в неонатальном периоде
Поздно появляющиеся острые
ВАРИАНТЫ КЛИНИЧЕСКОЙ МАНИФЕСТАЦИИ НБО
Острые тяжелые расстройства в неонатальном периоде
Поздно появляющиеся острые

НБО С ПСИХИАТРИЧЕСКИМИ И ПОВЕДЕНЧЕСКИМИ РАССТРОЙСТВАМИ
НБО С ПСИХИАТРИЧЕСКИМИ И ПОВЕДЕНЧЕСКИМИ РАССТРОЙСТВАМИ

ЭТАПЫ ДИАГНОСТИКИ РЕДКИХ НБО
Стандартные скрининговые тесты
Количественные исследования
Нагрузочные пробы
Исследования вовлеченности в патологический
ЭТАПЫ ДИАГНОСТИКИ РЕДКИХ НБО
Стандартные скрининговые тесты
Количественные исследования
Нагрузочные пробы
Исследования вовлеченности в патологический

СПИСОК НБО,
ПОДЛЕЖАЩИХ ДНК-ДИАГНОСТИКЕ
- Адреногенитальный синдром
- Адренолейкодистрофия
- Альбинизм
- Болезнь Вильсона-Коновалова
- Болезнь
СПИСОК НБО,
ПОДЛЕЖАЩИХ ДНК-ДИАГНОСТИКЕ
- Адреногенитальный синдром
- Адренолейкодистрофия
- Альбинизм
- Болезнь Вильсона-Коновалова
- Болезнь

ПАТОМОРФОЛОГОГИЧЕСКИЕ ИССЛЕДОВАНИЯ
ПАТОМОРФОЛОГОГИЧЕСКИЕ ИССЛЕДОВАНИЯ

ПРИНЦИПЫ ЛЕЧЕНИЯ НБО
Контроль за накоплением субстрата
1.1. Диета с удалением субстрата
ПРИНЦИПЫ ЛЕЧЕНИЯ НБО
Контроль за накоплением субстрата
1.1. Диета с удалением субстрата

ДИЕТА С УДАЛЕНИЕМ СУБСТРАТА
ДИЕТА С УДАЛЕНИЕМ СУБСТРАТА

УСКОРЕННОЕ ВЫВЕДЕНИЕ СУБСТРАТА
Диализ (перитонеальный диализ, гемодиализ, длительная вено-венозная гемофильтрация)
Использование бензоата, фенилацетата
УСКОРЕННОЕ ВЫВЕДЕНИЕ СУБСТРАТА
Диализ (перитонеальный диализ, гемодиализ, длительная вено-венозная гемофильтрация)
Использование бензоата, фенилацетата

КОНТРОЛЬ ЗА ЭНДОГЕННОЙ ПРОДУКЦИЕЙ СУБСТРАТА
Болезни аминокислотного обмена и дефекты цикла
КОНТРОЛЬ ЗА ЭНДОГЕННОЙ ПРОДУКЦИЕЙ СУБСТРАТА
Болезни аминокислотного обмена и дефекты цикла

ПРИМЕРЫ КОФАКТОРОВ, ИСПОЛЬЗУЕМЫХ ПРИ ЛЕЧЕНИИ НЕКОТОРЫХ НБО
ПРИМЕРЫ КОФАКТОРОВ, ИСПОЛЬЗУЕМЫХ ПРИ ЛЕЧЕНИИ НЕКОТОРЫХ НБО

ЗАМЕСТИТЕЛЬНАЯ ФЕРМЕНТОТЕРАПИЯ
Болезнь Гоше – препарат церезим
ЗАМЕСТИТЕЛЬНАЯ ФЕРМЕНТОТЕРАПИЯ
Болезнь Гоше – препарат церезим

ПРИМЕРЫ ТРАСПЛАНТАЦИИ ОРГАНОВ
ПРИ НБО
Трансплантация печени:
Недостаточность α1-антитрипсина, наследственная тирозинемия,
ПРИМЕРЫ ТРАСПЛАНТАЦИИ ОРГАНОВ
ПРИ НБО
Трансплантация печени:
Недостаточность α1-антитрипсина, наследственная тирозинемия,

ПРИНЦИПЫ ИНДИВИДУАЛЬНОЙ,
СЕМЕЙНОЙ И МАССОВОЙ ПРОФИЛАКТИКИ НБО
1. Своевременная диагностика, лабораторное подтверждение
ПРИНЦИПЫ ИНДИВИДУАЛЬНОЙ,
СЕМЕЙНОЙ И МАССОВОЙ ПРОФИЛАКТИКИ НБО
1. Своевременная диагностика, лабораторное подтверждение

ПРИМЕРЫ
НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
ОБМЕНА ВЕЩЕСТВ
ПРИМЕРЫ
НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
ОБМЕНА ВЕЩЕСТВ

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ЛАКТАЗНАЯ НЕДОСТАТОЧНОСТЬ
Дефектый фермент: лактаза (дисахаридаза) расщепляет молочный сахар-лактозу на
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ЛАКТАЗНАЯ НЕДОСТАТОЧНОСТЬ
Дефектый фермент: лактаза (дисахаридаза) расщепляет молочный сахар-лактозу на

Генетически и клинически гетерогенная группа заболеваний
Дефектый фермент: галактокиназа - GALK, галактозо-1-фосфат-уридилтрансфераза
Дефектый фермент: галактокиназа - GALK, галактозо-1-фосфат-уридилтрансфераза

Мутантный ген – GALT на 9р13
Дефектный фермент - галактозо-1-фосфатуридилтрансфераза
Тип наследования: АР
Мутантный ген – GALT на 9р13
Дефектный фермент - галактозо-1-фосфатуридилтрансфераза
Тип наследования: АР

Вторичные осложнения: -спленомегалия, -нарушение свертывания крови, -картина геморрагического диатеза и гемолитической
Вторичные осложнения: -спленомегалия, -нарушение свертывания крови, -картина геморрагического диатеза и гемолитической

Критерии диагноза:
повышенные концентрации галактозы и/или галактозо-1-фосфата,
снижение содержания глюкозы и толерантности
Критерии диагноза:
повышенные концентрации галактозы и/или галактозо-1-фосфата,
снижение содержания глюкозы и толерантности
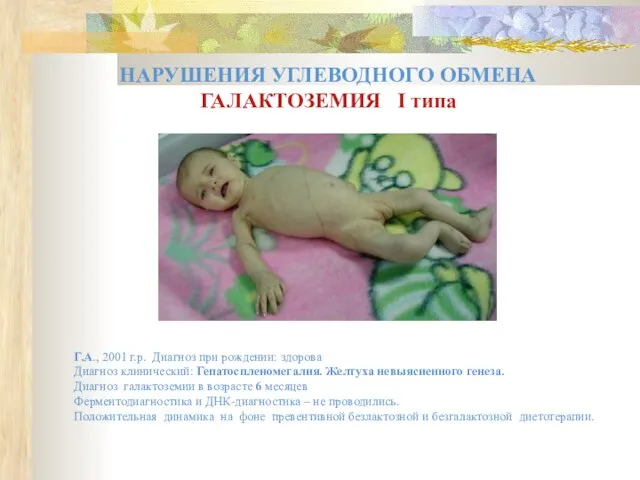
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГАЛАКТОЗЕМИЯ I типа
Г.А., 2001 г.р. Диагноз при рождении: здорова
Диагноз
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГАЛАКТОЗЕМИЯ I типа
Г.А., 2001 г.р. Диагноз при рождении: здорова
Диагноз

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГАЛАКТОЗЕМИЯ II типа
Мутантный ген – GALК на 17q24
Дефектный
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГАЛАКТОЗЕМИЯ II типа
Мутантный ген – GALК на 17q24
Дефектный

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГАЛАКТОЗЕМИЯ II типа
С.Э., 1998 г.р. Диагноз при рождении: ВУ
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГАЛАКТОЗЕМИЯ II типа
С.Э., 1998 г.р. Диагноз при рождении: ВУ

Мутантный ген – ALDA картирован на 9q21.3-22.2
Дефектый фермент - альдолаза
Мутантный ген – ALDA картирован на 9q21.3-22.2
Дефектый фермент - альдолаза

Клинические проявления
рвота
отвращение к пище содержащей фруктозу
гепатомегалия, желтуха
слабость, вялость
гипервозбудимость
судороги,
Клинические проявления
рвота
отвращение к пище содержащей фруктозу
гепатомегалия, желтуха
слабость, вялость
гипервозбудимость
судороги,

Лабораторные данные: -фруктозурия, -альбуминурия, -гипераминоацидурия, -гиперфруктоземия после нагрузки фруктозой, гипогликемия
Лечение: диета
Лабораторные данные: -фруктозурия, -альбуминурия, -гипераминоацидурия, -гиперфруктоземия после нагрузки фруктозой, гипогликемия
Лечение: диета

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ФРУКТОЗЕМИЯ
К.М., 1987 г.р. Диагноз при рождении: здоров. Болеет с
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ФРУКТОЗЕМИЯ
К.М., 1987 г.р. Диагноз при рождении: здоров. Болеет с

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГЛЮКОЦЕРЕБРОЗИДОЗЫ
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГЛЮКОЦЕРЕБРОЗИДОЗЫ

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
БОЛЕЗНЬ ГОШЕ
АР-наследование Ген GBA, 1q21-q31 Измененный фермент - глюкоцереброзидаза
Г.А.,
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
БОЛЕЗНЬ ГОШЕ
АР-наследование Ген GBA, 1q21-q31 Измененный фермент - глюкоцереброзидаза
Г.А.,

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
МУКОПОЛИСАХАРИДОЗЫ
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
МУКОПОЛИСАХАРИДОЗЫ

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
МУКОПОЛИСАХАРИДОЗ VI типа
АР-наследование Ген ARSB, 5q13.3 Измененный фермент -
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
МУКОПОЛИСАХАРИДОЗ VI типа
АР-наследование Ген ARSB, 5q13.3 Измененный фермент -

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГЛИКОГЕНОЗЫ
Тип I - болезнь Гирке (10 клинических форм)
Тип
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
ГЛИКОГЕНОЗЫ
Тип I - болезнь Гирке (10 клинических форм)
Тип

Болезни углеводного обмена
Гликогенозы - Тип I (болезнь Гирке)
Дефектый фермент: глюкозо-6-фосфотаза,
Болезни углеводного обмена
Гликогенозы - Тип I (болезнь Гирке)
Дефектый фермент: глюкозо-6-фосфотаза,

Болезни углеводного обмена
Гликогенозы - Тип I (болезнь Гирке)
Большой живот,
Болезни углеводного обмена
Гликогенозы - Тип I (болезнь Гирке)
Большой живот,

Болезни углеводного обмена
Гликогенозы - Тип II (болезнь Помпе)
Дефектый фермент: α-D-глюкозидаза
Болезни углеводного обмена
Гликогенозы - Тип II (болезнь Помпе)
Дефектый фермент: α-D-глюкозидаза

Болезни углеводного обмена
Гликогенозы - Тип II (болезнь Помпе)
Мышечная слабость, включая
Болезни углеводного обмена
Гликогенозы - Тип II (болезнь Помпе)
Мышечная слабость, включая

Болезни углеводного обмена
Гликогенозы - Тип III (болезнь Кори)
Дефектый фермент: амило-1,6-глюкозидаза,
Болезни углеводного обмена
Гликогенозы - Тип III (болезнь Кори)
Дефектый фермент: амило-1,6-глюкозидаза,

Болезни углеводного обмена
Гликогенозы - Тип III (болезнь Кори)
гепатомегалия
гипогликемия
гиперлипидемия
толерантность к глюкозе
Болезни углеводного обмена
Гликогенозы - Тип III (болезнь Кори)
гепатомегалия
гипогликемия
гиперлипидемия
толерантность к глюкозе

Болезни углеводного обмена
Гликогенозы - Тип IV (болезнь Андерсена)
Дефектый фермент: амило-1,4:1,6-глюкотрансфераза,
Болезни углеводного обмена
Гликогенозы - Тип IV (болезнь Андерсена)
Дефектый фермент: амило-1,4:1,6-глюкотрансфераза,

Болезни углеводного обмена
Гликогенозы - Тип IV (болезнь Андерсена)
Рвота
диарея
нарушение вскармливания
задержка физического
Болезни углеводного обмена
Гликогенозы - Тип IV (болезнь Андерсена)
Рвота
диарея
нарушение вскармливания
задержка физического

Болезни углеводного обмена
Гликогенозы - Тип V (болезнь Мак-Ардла)
Дефектый фермент: мышечная
Болезни углеводного обмена
Гликогенозы - Тип V (болезнь Мак-Ардла)
Дефектый фермент: мышечная

Болезни углеводного обмена
Гликогенозы - Тип VI (болезнь Герса)
Дефектный фермент: фосфорилаза
Тип
Болезни углеводного обмена
Гликогенозы - Тип VI (болезнь Герса)
Дефектный фермент: фосфорилаза
Тип

Болезни обмена аминокислот
Фенилкетонурия I, II, III типы, материнская ФКУ
Тирозинемия I, II,
Болезни обмена аминокислот
Фенилкетонурия I, II, III типы, материнская ФКУ
Тирозинемия I, II,





НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ
ФЕНИЛКЕТОНУРИЯ
С.А., 1983 г.р.; Диагноз ФКУ в 2,5 года (г.Санкт-петербург);
НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ
ФЕНИЛКЕТОНУРИЯ
С.А., 1983 г.р.; Диагноз ФКУ в 2,5 года (г.Санкт-петербург);

НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ ТИРОЗИНЕМИЯ
АР-наследование Ген FAH - 15q23-25 Измененный фермент -
НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ ТИРОЗИНЕМИЯ
АР-наследование Ген FAH - 15q23-25 Измененный фермент -

НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ
АЛЬБИНИЗМ
АР-наследование Ген TYR - 11q14-q21 Измененный фермент –
НАРУШЕНИЯ ОБМЕНА АМИНОКИСЛОТ
АЛЬБИНИЗМ
АР-наследование Ген TYR - 11q14-q21 Измененный фермент –
 Аффективные расстройства
Аффективные расстройства Коронавирус. Профилактика коронавирусной инфекции
Коронавирус. Профилактика коронавирусной инфекции Обморожение. Первая помощь при обморожении
Обморожение. Первая помощь при обморожении Осложнения индукции родов. Механизм их развития с последующими путями коррекции
Осложнения индукции родов. Механизм их развития с последующими путями коррекции Цирроз печени: актуальность, патогенез, синдромы, пути терапии
Цирроз печени: актуальность, патогенез, синдромы, пути терапии Клиническая фармакология средств, используемых при спазме бронхиальной мускулатуры
Клиническая фармакология средств, используемых при спазме бронхиальной мускулатуры Шизофрения. Диагностика,емі. Шизофренияның заманауи теориялары
Шизофрения. Диагностика,емі. Шизофренияның заманауи теориялары Ринопластика
Ринопластика Логопедический массаж и самомассаж как здоровьесберегающая технология
Логопедический массаж и самомассаж как здоровьесберегающая технология Гломерулонефриты
Гломерулонефриты Любовь, созависимость и другие типы эмоциональной зависимости
Любовь, созависимость и другие типы эмоциональной зависимости Синдром Эдвардса
Синдром Эдвардса Средства, влияющие на афферентную иннервацию
Средства, влияющие на афферентную иннервацию Микрохирургическая бригада
Микрохирургическая бригада Недоверие и страх системы ОСМС
Недоверие и страх системы ОСМС Ретенция результатов лечения аномалий зубочелюстной системы. Рецидивы
Ретенция результатов лечения аномалий зубочелюстной системы. Рецидивы Лекция по ЭКГ вкратце
Лекция по ЭКГ вкратце Естественная резистентность
Естественная резистентность Анатомофизиологический обзор центральной нервной системы
Анатомофизиологический обзор центральной нервной системы Игра в дошкольном и младшем школьном возрасте
Игра в дошкольном и младшем школьном возрасте Пункционные малоинвазивные вмешательства под контролем ультразвуковой томографии органов брюшной полости. Основные понятия
Пункционные малоинвазивные вмешательства под контролем ультразвуковой томографии органов брюшной полости. Основные понятия Презентация по медицине Туберкулёз
Презентация по медицине Туберкулёз  Дерматологические аспекты ВИЧ-инфекции
Дерматологические аспекты ВИЧ-инфекции Практикум по методам психологического исследования. Часть 2
Практикум по методам психологического исследования. Часть 2 Оптические иллюзии и их коррекции
Оптические иллюзии и их коррекции Аттестационная работа. Влияние на организм младшего школьника компьютерных и подвижных игр
Аттестационная работа. Влияние на организм младшего школьника компьютерных и подвижных игр Дезинфекция термометров, тонометров
Дезинфекция термометров, тонометров Острые стрессовые реакции. Работа с пострадавшими
Острые стрессовые реакции. Работа с пострадавшими