- Наследственные заболевания и генетические синдромы

Содержание
- 2. Наследственные болезни возникают в результате мутаций наследственных структур –– хромосом или генов. Соответственно выделяют хромосомные и
- 3. РОЛЬ ОТДЕЛЬНЫХ ФАКТОРОВ В РАЗВИТИИ ВРОЖДЕННЫХ И НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ У ДЕТЕЙ
- 4. Классификация мутаций по месту их возникновения
- 5. Генеративные В половых клетках Моногенные Хромосомные Полигенные Митохонд-риальные
- 6. Классификация мутаций по характеру проявления
- 7. Генные болезни
- 8. 1. Аутосомно-доминантные моногенные болезни Действие мутантного гена проявляется практически всегда Больные мальчики и девочки рождаются с
- 9. ПОЛИДАКТИЛИЯ Клинические признаки: существует два варианта: тип А, при котором дополнительный палец функционален, и тип В,
- 10. ОСТЕОГЕНЕЗ Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключиц при минимальной травме, деформации конечностей, голубые
- 11. СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз) Синдром Крузона – дефект гена каспазы, 10q. Впервые описан в 1912 г.
- 12. ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ») Клинические признаки: чрезмерный рост волос на всех частях тела, кроме ладоней и
- 13. Аутосомно-рецессивные моногенные болезни Мутантный ген проявляется только в гомозиготном состоянии, а гетерозиготное состояние -, так называемый,
- 14. РАСЩЕЛИНА ГУБЫ Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и телоризм, деформации первых пальцев
- 15. ЧЕРЕП В ФОРМЕ ТРИЛИСТНИКА Клинические признаки: характерная форма черепа (возникает вследствие внутриутробного зарастания швов) и лица,
- 16. ФЕНИЛКЕТОНУРИЯ Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом. Патология связана с недостаточностью
- 17. МУКОПОЛИСАХАРИДОЗ Синдром Моркио описан в 1929 г. Клинические признаки: отставание в росте, деформация позвоночника и грудины,
- 18. Ихтиоз Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся нарушением ороговения, проявляется образованием на коже чешуек,
- 19. Прогерия Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений кожи, внутренних органов, обусловленных
- 20. 3. ХРОМОСОМНЫЕ БОЛЕЗНИ Хромосомные заболевания связаны с аномалиями числа или структуры хромосом. Для них характерно: малый
- 21. Генеративные мутации ХХУ; ХУУ- синдром Клайнфельтера. ( лишняя Х у мужчин - ХХУ) ХО- синдром Шершевского-
- 22. СИНДРОМ ДАУНА (ТРИСОМИЯ 21) Описан в 1866 г. Клинические признаки: умственная отсталость, плоское лицо, монголоид -ный
- 23. СИНДРОМ ПАТАУ (ТРИСОМИЯ 13) Описан в 1961 г. Клинические признаки: микроцефалия, расщепле- ние губы и неба,
- 24. СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ) Описан в 1942 г. Клинические признаки: высокий рост, хрупкое телосложение, гипоплазия яичек,
- 25. СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ) Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые вторичные половые признаки, крыловидные
- 26. СИНДРОМ КОШАЧЬЕГО КРИКА (МОНОСОМИЯ 5р) Описан в 1963 г. Клинические признаки: необычный плач, напоминающий кошачье мяуканье,
- 27. Синдром трисомии 9р Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез глаз, глубоко посаженные глаза
- 28. Заболевания обусловлены полимерным характером взаимодействия генов или сочетанием взаимодействия нескольких генов и факторов среды (мультифакториальные заболевания).
- 29. В ДНК митохондрий 37 генов, они участвуют в выработке энергии, следовательно заболевания, связанные с мутациями в
- 30. В Женской консультации: Встать на учет в женской консультации как можно раньше! Оптимально – 6-10 недель
- 32. Скачать презентацию
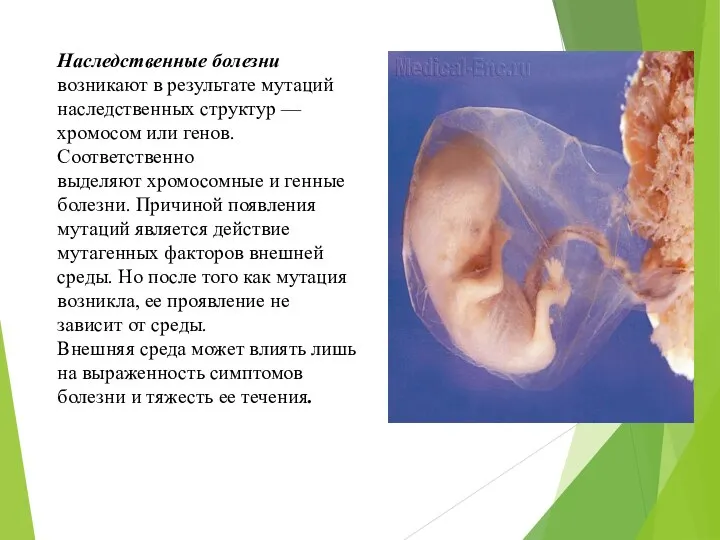
Наследственные болезни возникают в результате мутаций
наследственных структур –– хромосом или генов.
Наследственные болезни возникают в результате мутаций
наследственных структур –– хромосом или генов.

РОЛЬ ОТДЕЛЬНЫХ ФАКТОРОВ В РАЗВИТИИ
ВРОЖДЕННЫХ И НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ У ДЕТЕЙ
РОЛЬ ОТДЕЛЬНЫХ ФАКТОРОВ В РАЗВИТИИ
ВРОЖДЕННЫХ И НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ У ДЕТЕЙ
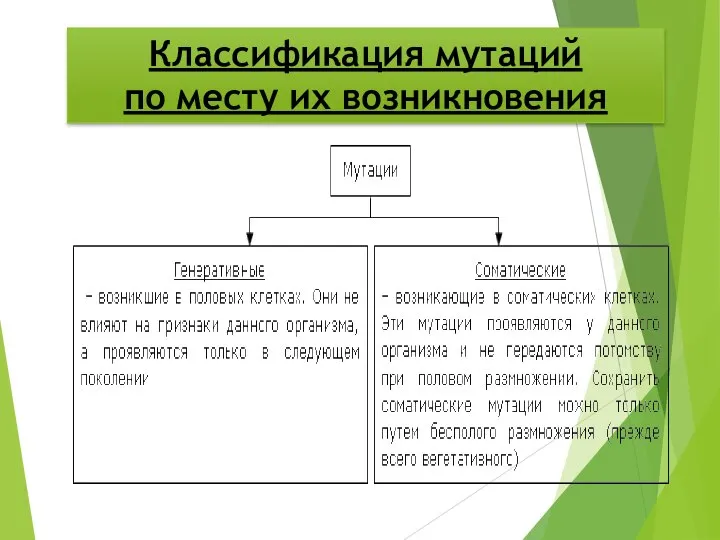
Классификация мутаций
по месту их возникновения
Классификация мутаций
по месту их возникновения
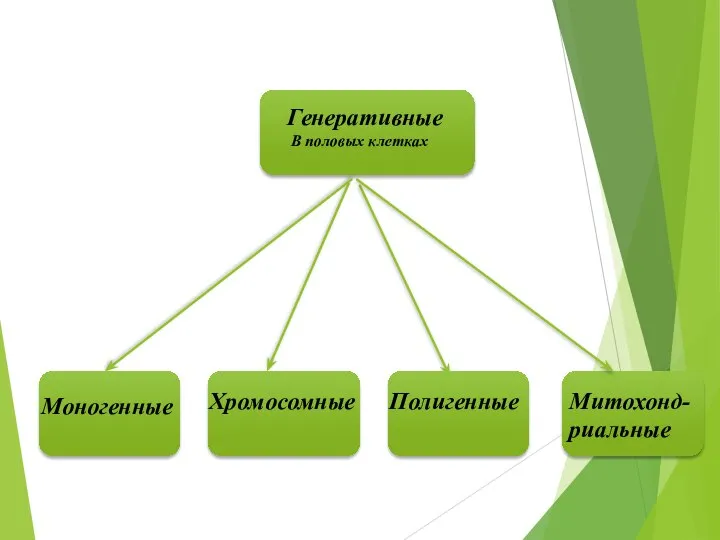
Генеративные
В половых клетках
Моногенные
Хромосомные
Полигенные
Митохонд-риальные
Генеративные
В половых клетках
Моногенные
Хромосомные
Полигенные
Митохонд-риальные
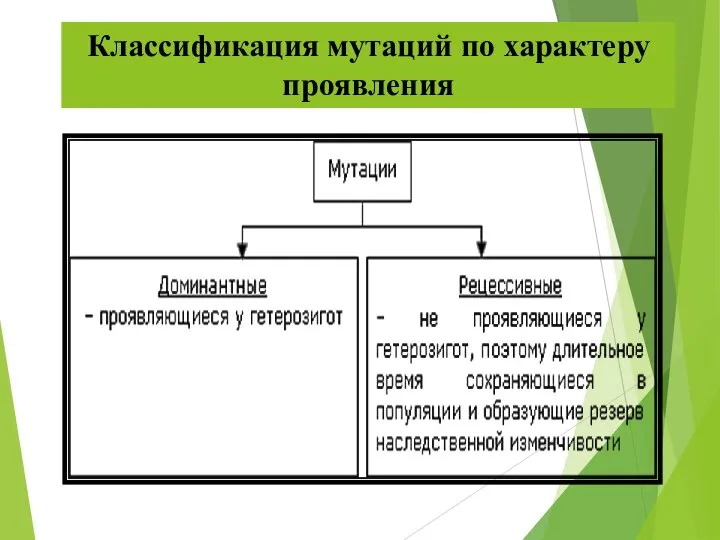
Классификация мутаций по характеру проявления
Классификация мутаций по характеру проявления
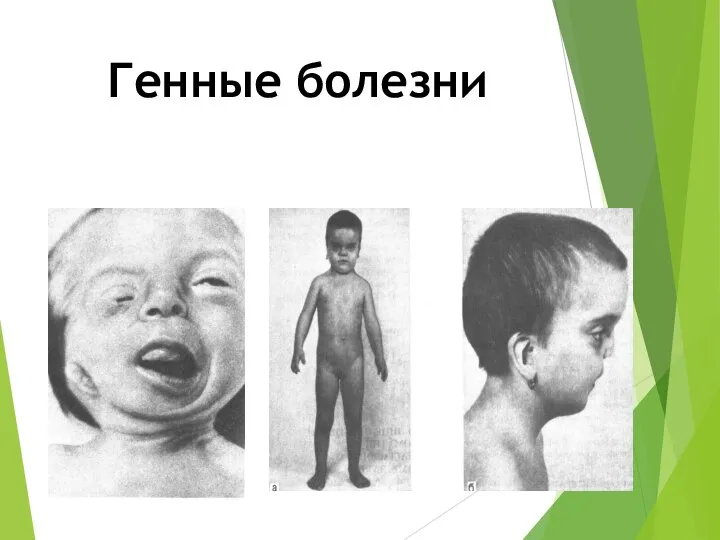
Генные болезни
Генные болезни

1. Аутосомно-доминантные
моногенные болезни
Действие мутантного гена проявляется практически всегда
Больные мальчики и
1. Аутосомно-доминантные
моногенные болезни
Действие мутантного гена проявляется практически всегда
Больные мальчики и

ПОЛИДАКТИЛИЯ
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
ПОЛИДАКТИЛИЯ
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
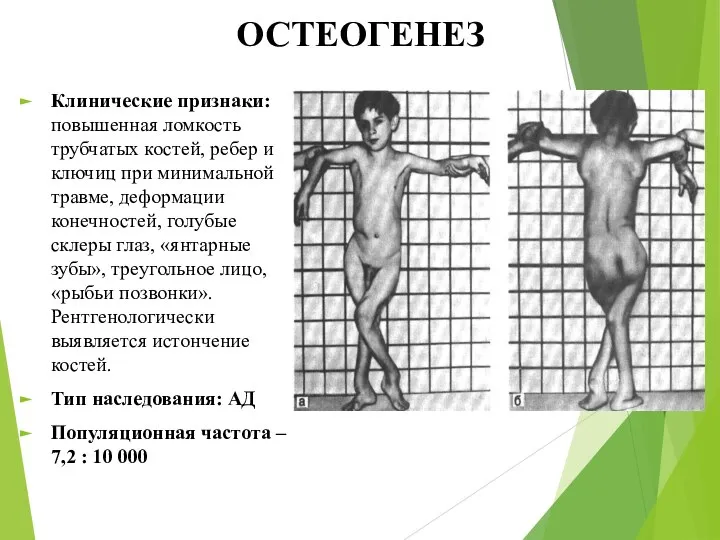
ОСТЕОГЕНЕЗ
Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключиц при минимальной
ОСТЕОГЕНЕЗ
Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключиц при минимальной
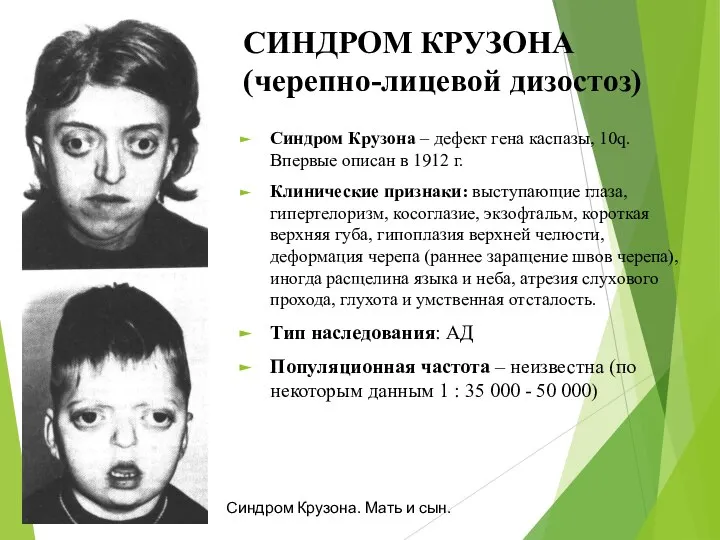
СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз)
Синдром Крузона – дефект гена каспазы, 10q. Впервые
СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз)
Синдром Крузона – дефект гена каспазы, 10q. Впервые

ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ»)
Клинические признаки: чрезмерный рост волос на всех частях
ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ»)
Клинические признаки: чрезмерный рост волос на всех частях

Аутосомно-рецессивные моногенные болезни
Мутантный ген проявляется только в гомозиготном состоянии,
Аутосомно-рецессивные моногенные болезни
Мутантный ген проявляется только в гомозиготном состоянии,
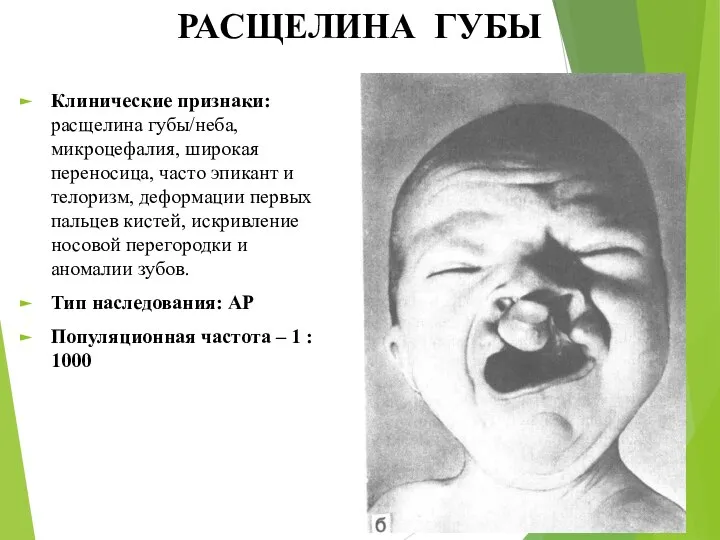
РАСЩЕЛИНА ГУБЫ
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и
РАСЩЕЛИНА ГУБЫ
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и

ЧЕРЕП В ФОРМЕ ТРИЛИСТНИКА
Клинические признаки: характерная форма черепа (возникает вследствие внутриутробного
ЧЕРЕП В ФОРМЕ ТРИЛИСТНИКА
Клинические признаки: характерная форма черепа (возникает вследствие внутриутробного

ФЕНИЛКЕТОНУРИЯ
Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом.
ФЕНИЛКЕТОНУРИЯ
Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом.
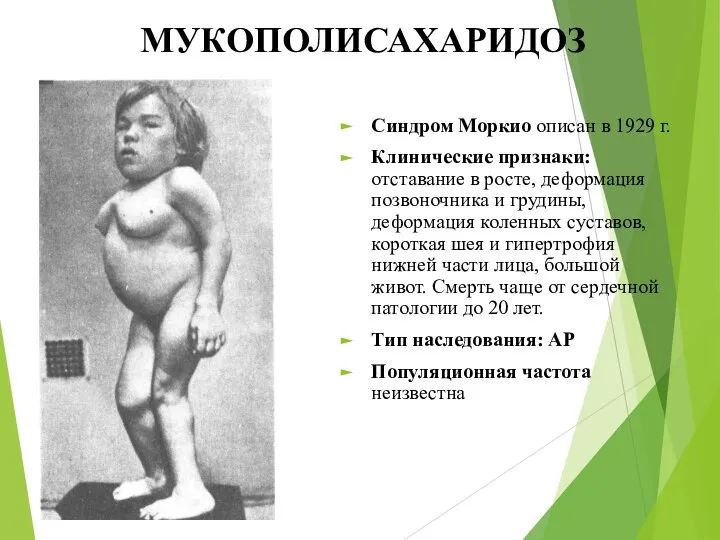
МУКОПОЛИСАХАРИДОЗ
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация
МУКОПОЛИСАХАРИДОЗ
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация
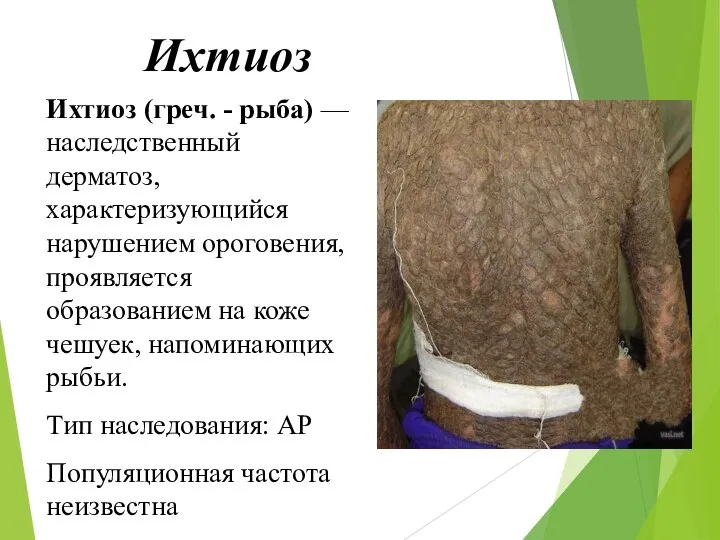
Ихтиоз
Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся нарушением ороговения, проявляется образованием
Ихтиоз
Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся нарушением ороговения, проявляется образованием

Прогерия
Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом
Прогерия
Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом

3. ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные заболевания связаны с аномалиями числа или структуры хромосом.
Для
3. ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные заболевания связаны с аномалиями числа или структуры хромосом.
Для

Генеративные мутации
ХХУ; ХУУ- синдром Клайнфельтера. ( лишняя Х у мужчин -
Генеративные мутации
ХХУ; ХУУ- синдром Клайнфельтера. ( лишняя Х у мужчин -

СИНДРОМ ДАУНА (ТРИСОМИЯ 21)
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское
СИНДРОМ ДАУНА (ТРИСОМИЯ 21)
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское
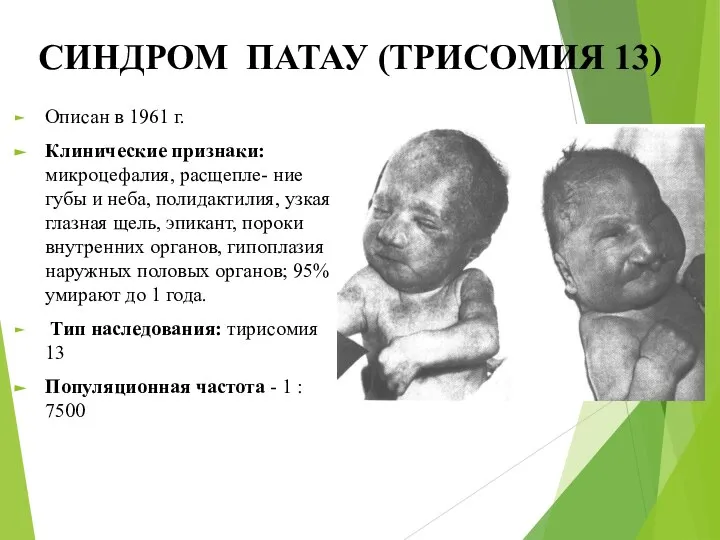
СИНДРОМ ПАТАУ (ТРИСОМИЯ 13)
Описан в 1961 г.
Клинические признаки: микроцефалия, расщепле- ние
СИНДРОМ ПАТАУ (ТРИСОМИЯ 13)
Описан в 1961 г.
Клинические признаки: микроцефалия, расщепле- ние

СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ)
Описан в 1942 г.
Клинические признаки: высокий рост, хрупкое
СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ)
Описан в 1942 г.
Клинические признаки: высокий рост, хрупкое
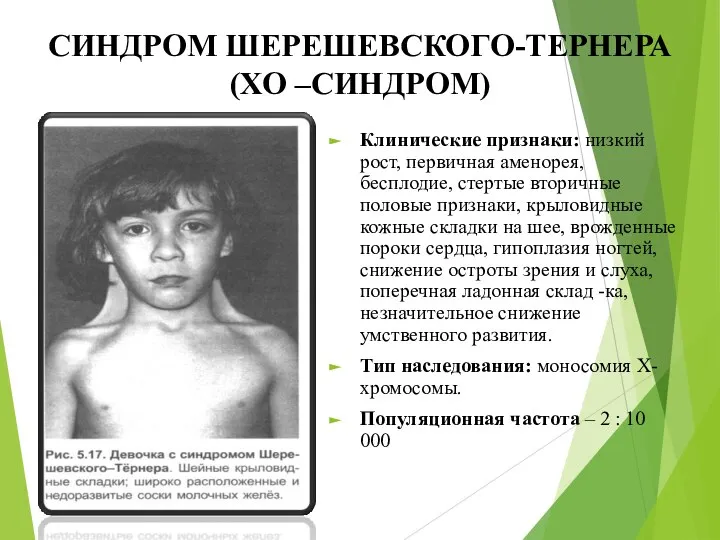
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ)
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ)
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые
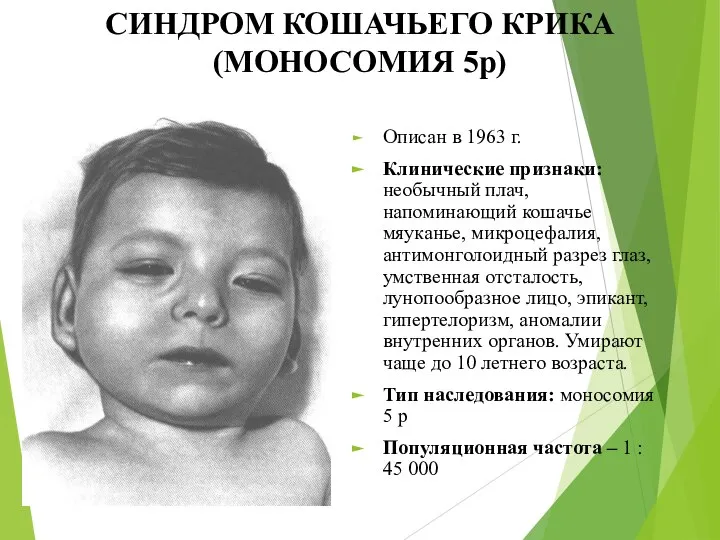
СИНДРОМ КОШАЧЬЕГО КРИКА (МОНОСОМИЯ 5р)
Описан в 1963 г.
Клинические признаки: необычный плач,
СИНДРОМ КОШАЧЬЕГО КРИКА (МОНОСОМИЯ 5р)
Описан в 1963 г.
Клинические признаки: необычный плач,
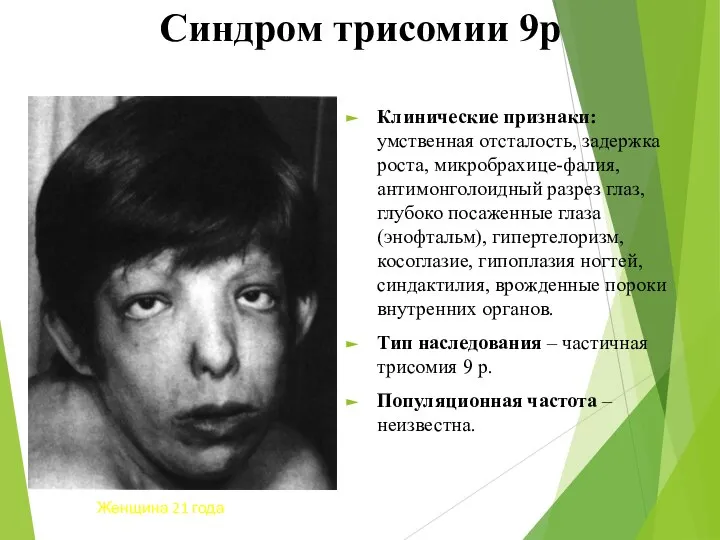
Синдром трисомии 9р
Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез
Синдром трисомии 9р
Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез
Заболевания обусловлены полимерным характером взаимодействия генов или сочетанием взаимодействия нескольких генов
Заболевания обусловлены полимерным характером взаимодействия генов или сочетанием взаимодействия нескольких генов

В ДНК митохондрий 37 генов, они участвуют в выработке энергии, следовательно
В ДНК митохондрий 37 генов, они участвуют в выработке энергии, следовательно
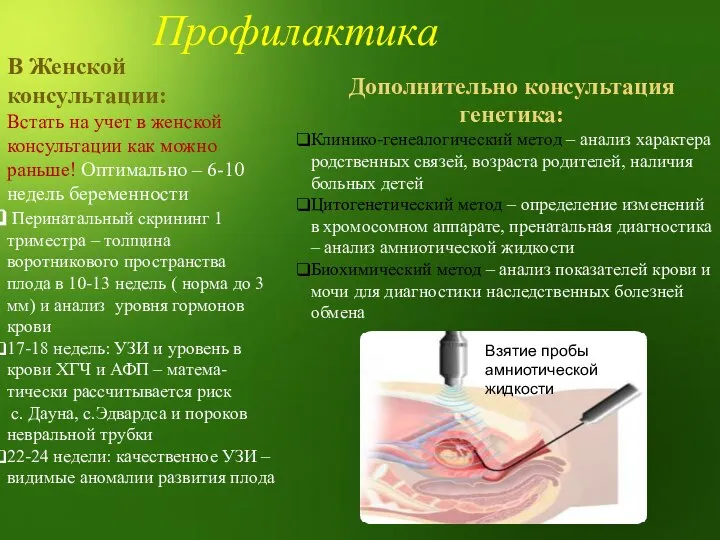
В Женской консультации:
Встать на учет в женской консультации как можно раньше!
В Женской консультации:
Встать на учет в женской консультации как можно раньше!
 Орфанные заболевания
Орфанные заболевания Опухоли опорно-двигательной системы
Опухоли опорно-двигательной системы Строение нефрона. Образование мочи
Строение нефрона. Образование мочи Электромагниттердің адам өміріне зияны
Электромагниттердің адам өміріне зияны Антиангинальные средства
Антиангинальные средства Первичная профилактика ВПЧ
Первичная профилактика ВПЧ Типичные ошибки семейного воспитания
Типичные ошибки семейного воспитания Синдром марфана
Синдром марфана Клини́ческая фармаколо́гия
Клини́ческая фармаколо́гия Коронарна хвороба cерця (ІХС). Стенокардія
Коронарна хвороба cерця (ІХС). Стенокардія Система здравоохранения в Российской Федерации
Система здравоохранения в Российской Федерации СРС. Круп при острых инфекциях у детей
СРС. Круп при острых инфекциях у детей Понятие о хирургии. Понятие о травмах и травматизме. Особенности травматизма у разных видов животных
Понятие о хирургии. Понятие о травмах и травматизме. Особенности травматизма у разных видов животных Сортировка и концепция АВС
Сортировка и концепция АВС Бронхиальная астма
Бронхиальная астма Симптоматическая артериальная гипертензия
Симптоматическая артериальная гипертензия Cтимуляторы центральной нервной системы
Cтимуляторы центральной нервной системы Общие принципы оказания первой медицинской помощи. Профилактика ВБИ
Общие принципы оказания первой медицинской помощи. Профилактика ВБИ Пороки развития уха
Пороки развития уха Введение в ветеринарию
Введение в ветеринарию Конусно-лучевая компьютерная томография в эндодонтии
Конусно-лучевая компьютерная томография в эндодонтии Фармакогнозия. Лекарственное сырье растительного и животного происхождения
Фармакогнозия. Лекарственное сырье растительного и животного происхождения Гломерулонефрит у детей
Гломерулонефрит у детей Планирование выполнения криомодификации операции Лабиринт III в сочетании с радиочастотной аблацией правого предсердия
Планирование выполнения криомодификации операции Лабиринт III в сочетании с радиочастотной аблацией правого предсердия Наследственные болезни человека
Наследственные болезни человека Кейбір сыртқы орта жайттарының организмге ауру туындатушы әсері
Кейбір сыртқы орта жайттарының организмге ауру туындатушы әсері Критерии психического здоровья и психического расстройства
Критерии психического здоровья и психического расстройства Гемобластози. Причини, клініка, діагностика, догляд, профілактика
Гемобластози. Причини, клініка, діагностика, догляд, профілактика