- Генетика человека

Содержание
- 2. Генетика человека Генетика человека изучает наследственность и изменчивость человека и, наряду с другими фундаментальными науками, является
- 3. Генетический материал человека хромосомная ДНК (95%) митохондриальная ДНК (5%) Присутствуют: кольцевые молекулы ДНК транспозируемые генетические элементы
- 4. Фракции хромосомной ДНК ДНК, кодирующая последовательности аминокислот в белках, ДНК, кодирующая рРНК, ДНК, кодирующая тРНК, ДНК,
- 5. Избыточная ДНК вставочные последовательности и участки между генами, интроны, многокопийные гены, неработающие гены, псевдогены, процессированные псевдогены,
- 6. Кариотип человека В 1956 г. практически одновременно две пары исследователей: Дж.Тио и А.Леван, Ч.Форд и Дж.Хаммертон
- 7. А А
- 9. Каждая хромосома и ее плечи занимают в ядре свою собственную территорию, не деля ее с другими
- 10. Вероятно, такое расположение R- и G-бэндов сохраняется и в митотических хромосомах. Существует гипотеза, согласно которой необходимый
- 11. G-бэнды - синий цвет, R-бэнды - зеленый цвет
- 12. При дифференциальном окрашивании в гомологичных хромосомах обнаружены различия в ширине некоторых полос. Эти полиморфные участки –
- 13. Хромосомы человека
- 14. Хромосома 1 Самая большая по длине. Содержит больше всех хромосом генетической информации о структуре человеческого организма
- 15. Хромосома 2 Количество генов – 2500. Количество оснований – более 240 млн, из которых 95% определены.
- 16. Хромосома 5 Количество генов – 1700. Количество оснований – около 180 млн, из которых 95% определены.
- 17. Хромосома 11 Количество генов – 2000. Количество оснований – более 130 млн, из которых 95% определены.
- 18. Хромосома 22 Самая маленькая хромосома – 1,6 – 1,8% генома. Более 40 млн оснований, из которых
- 19. Основные часто встречающиеся последовательности – Alu-повторы, которые формируют блоки в районе центромеры и почти в центре
- 22. Половые хромосомы
- 23. Х- и Y-хромосомы совершенно разные, кроме псевдоаутосомных участков. Псевдоаутосомные участки: располагаются на концах р плеч, фактически
- 24. Х-хромосома Одна из самых больших, 5% от общей длины генома (155 миллионов химических звеньев ДНК), 1098
- 25. инактивация – дозовая компенсация, происходит в эмбриональном периоде, центр инактивации – Xic расположен в q-плече хромосомы,
- 26. Y-хромосома Более 50 млн п.н., 6 млн – палиндромы. Содержит 78 генов (большинство генов располагается в
- 27. Имеет место «перетасовка» генов, что: способствует самовосстановлению и элиминации мутантных генов, приводит к потере нормальных генов
- 28. Генетический материал стабилен, но возможны перестройки
- 29. Основа патологии – изменчивость
- 30. Каждая форма изменчивости может привести к развитию болезни
- 31. Мутации могут: элиминироваться из генофонда популяции (т.к. клетки и организмы оказываются нежизнеспособными, или организмы - стерильными)
- 32. Хромосомные мутации Изменения числа хромосом Гаплоидия Полиплоидия Гетероплоидия Трисомия Моносомия Нулисомия Изменения структуры хромосом Делеция Дупликация
- 33. Трисомии по 8 и 22 парам
- 34. Моносомия 45,Х
- 35. Делеция 22р-
- 36. Дупликация
- 37. Слияние хромосом Робертсоновские транслокации 14 21 14 21
- 38. 14 21 14+21 14,21 14+21 21 14+21,14 14+21,21 14 14,21 14,21 14,21 14+21 14,21 14+21,21 14,21
- 39. Кольцевая хромосома (17)
- 40. Изохромосомы p p p q q q q p p p q p q q p
- 41. Формирование однородительской дисомии
- 42. Наследственность и патология Причина наследственной патологии – наследственная изменчивость Мутационный процесс приводит к: социальной дезадаптации, снижению
- 43. Классификация болезней Наследственные болезни Болезни с наследственной предрасположенностью (бÓльшее значение имеет генетический материал) Болезни с наследственной
- 44. Наследственные болезни Теоретически число наследственных болезней может составлять 50 000 – 100 000 Виды наследственной патологии:
- 45. Классификация наследственных болезней Генетическая Клиническая Патогенетическая
- 46. Генетическая классификация хромосомные болезни, генные болезни, болезни с предрасположением (моногенные и полигенные), генетические болезни соматических клеток,
- 47. Выделяют также: митохондриальные болезни, болезни импринтинга, эпигенетические болезни, болезни экспансии тринуклеотидных повторов, прионные болезни.
- 48. Хромосомные болезни Причина – хромосомные мутации. Типы генетических эффектов при хромосомных нарушениях: специфические, полуспецифические, неспецифические.
- 50. Фенотипическое проявление хромосомных мутаций зависит от: типа мутации, типа хромосомы, типа поврежденного сегмента и его размеров,
- 51. Классификация хромосомных болезней Болезни, связанные с изменением числа хромосом. Болезни, обусловленные изменением структуры хромосом.
- 52. При классификации хромосомных болезней необходимо учитывать следующие моменты: а) характеристика мутации (полисомия, трисомия, моносомия, делеция, транслокация
- 53. Для точной диагностики хромосомных болезней определяют: тип мутации, мутантную хромосому, форму болезни (полная или мозаичная), вид
- 54. Синдром Дауна (47,XX+21 или 47,XY+21) Кариотип при синдроме Дауна
- 55. Частота 1:750 новорождённых. Характерна малая средняя продолжительность жизни (35 лет). Цитогенетические варианты: полная трисомия (90-95% всех
- 59. Синдром Патау (47,XX+13 или 47,XY+13) Кариотип при синдроме Патау
- 60. Частота – 1:6000 Соотношение полов 1:1 Цитогенетические варианты: полная трисомия, мозаицизм, робертсоновская транслокация, изохромосомы.
- 61. Фенотипические проявления: сниженная масса тела, микроцефалия, недоразвитие мозга, аномалии лица (запавшая переносица, расщелина верхней губы и
- 64. Синдром Эдвардса (47,XX+18 или 47,XY+18) Кариотип при синдроме Эдвардса
- 65. Частота 1:5000 – 1:7000 Соотношение частоты заболевания у девочек и мальчиков составляет 3:1 Цитогенетические варианты: полная
- 66. Фенотипические проявления: Врожденные пороки развития: лицевого черепа, сердца (дефект межжелудочковой перегородки и открытый боталлов проток), костной
- 70. Синдром Шерешевского-Тернера (45,X) Частота 1:2000 – 1:5000 девочек Цитогенетические варианты: полная моносомия 45,Х (50% от всех
- 71. Основные клинические признаки: нанизм, крыловидные кожные складки на шее, короткая шея с низкой линией роста волос,
- 74. Синдром трисомии 8 (47,XX+8 или 47,XY+8) Клиническая картина синдрома впервые описана разными авторами в 1962 и
- 75. Основные клинические признаки: отставание в умственном развитии, отклонения в строении лица (выступающий лоб, косоглазие глубоко посаженные
- 79. Синдром трисомии 14 (47,XX+14 или 47,XY+14) Описан в 1975 году. Прогноз жизни неблагоприятный, но отмечены больные
- 80. Основные клинические признаки: микроцефалия, асимметрия лица, высокий и выступающий лоб, нос короткий и бульбообразный, губы полные,
- 81. Синдроме Клайнфельтера (47,XXY, 48,XXXY и т.д.) Кариотип при синдроме Клайнфельтера
- 82. Частота 1:1000 мальчиков (1:500 – 1:750) Цитогенетические варианты: 80% - ХХY, 20% - мозаицизм; возможно 48,ХХХY.
- 83. Синдром Лежена - синдром кошачьего крика (46,XX5p- или 46,XY5p-) Кариотип при синдроме Лежена
- 84. Частота 1:45000 новорожденных Цитогенетические варианты: образование кольцевой хромосомы 5 с частичной потерей плеча; делеция 1/2 или1/3
- 85. Основные клинические признаки: нарушения гортани и специфический плач, микроцефалия, умственная отсталость, лунообразное лицо, низко расположенные деформированные
- 88. Синдром Вольфа-Хиршхорна (46,XX4p- или 46,XY4p-) Частота 1:100 000 Жизнеспособность детей резко снижена, большинство умирают в возрасте
- 89. Основные клинические признаки: более чем 50% детей имеют пороки внутренних органов (сердца, почек, ЖКТ), врожденные пороки
- 91. Ретинобластома (микроцитогенетический синдром 13q-14) (46,XX13q- или 46,XY13q-) Частота 1:15 000 – 1:34 000 новорождённых Болезнь с
- 93. Скачать презентацию

Генетика человека
Генетика человека изучает наследственность и изменчивость человека и, наряду с
Генетика человека
Генетика человека изучает наследственность и изменчивость человека и, наряду с

Генетический материал человека
хромосомная ДНК (95%)
митохондриальная ДНК (5%)
Присутствуют:
кольцевые молекулы ДНК
транспозируемые генетические
Генетический материал человека
хромосомная ДНК (95%)
митохондриальная ДНК (5%)
Присутствуют:
кольцевые молекулы ДНК
транспозируемые генетические

Фракции хромосомной ДНК
ДНК, кодирующая последовательности аминокислот в белках,
ДНК, кодирующая рРНК,
ДНК, кодирующая
Фракции хромосомной ДНК
ДНК, кодирующая последовательности аминокислот в белках,
ДНК, кодирующая рРНК,
ДНК, кодирующая

Избыточная ДНК
вставочные последовательности и участки между генами,
интроны,
многокопийные гены,
неработающие гены,
псевдогены,
процессированные псевдогены,
повторы,
сателлитная ДНК.
Избыточная ДНК
вставочные последовательности и участки между генами,
интроны,
многокопийные гены,
неработающие гены,
псевдогены,
процессированные псевдогены,
повторы,
сателлитная ДНК.

Кариотип человека
В 1956 г. практически одновременно две пары исследователей: Дж.Тио и А.Леван,
Кариотип человека
В 1956 г. практически одновременно две пары исследователей: Дж.Тио и А.Леван,

А
А
А
А


Каждая хромосома и ее плечи занимают в ядре свою собственную территорию,
Каждая хромосома и ее плечи занимают в ядре свою собственную территорию,

Вероятно, такое расположение R- и G-бэндов сохраняется и в митотических хромосомах.
Существует
Вероятно, такое расположение R- и G-бэндов сохраняется и в митотических хромосомах.
Существует

G-бэнды - синий цвет, R-бэнды - зеленый цвет
G-бэнды - синий цвет, R-бэнды - зеленый цвет

При дифференциальном окрашивании в гомологичных хромосомах обнаружены различия в ширине некоторых
При дифференциальном окрашивании в гомологичных хромосомах обнаружены различия в ширине некоторых

Хромосомы человека
Хромосомы человека

Хромосома 1
Самая большая по длине.
Содержит больше всех хромосом генетической информации о
Хромосома 1
Самая большая по длине.
Содержит больше всех хромосом генетической информации о

Хромосома 2
Количество генов – 2500.
Количество оснований – более 240 млн, из
Хромосома 2
Количество генов – 2500.
Количество оснований – более 240 млн, из

Хромосома 5
Количество генов – 1700.
Количество оснований – около 180 млн, из
Хромосома 5
Количество генов – 1700.
Количество оснований – около 180 млн, из

Хромосома 11
Количество генов – 2000.
Количество оснований – более 130 млн, из
Хромосома 11
Количество генов – 2000.
Количество оснований – более 130 млн, из

Хромосома 22
Самая маленькая хромосома – 1,6 – 1,8% генома.
Более 40 млн
Хромосома 22
Самая маленькая хромосома – 1,6 – 1,8% генома.
Более 40 млн

Основные часто встречающиеся последовательности – Alu-повторы, которые формируют блоки в районе
Основные часто встречающиеся последовательности – Alu-повторы, которые формируют блоки в районе

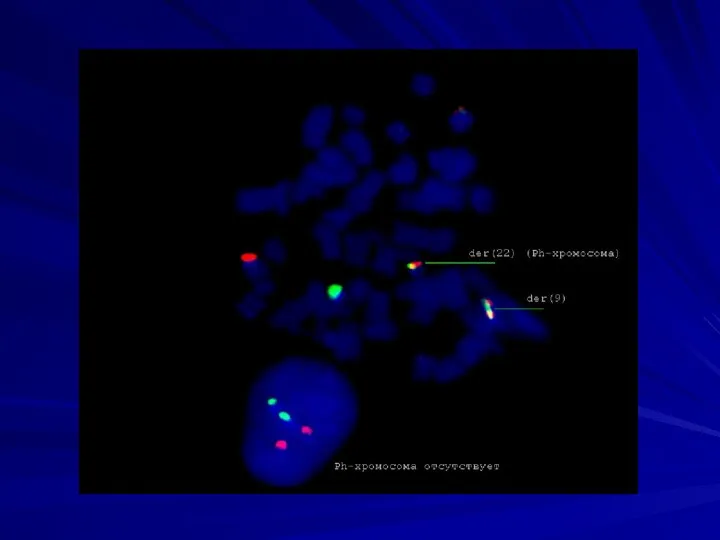
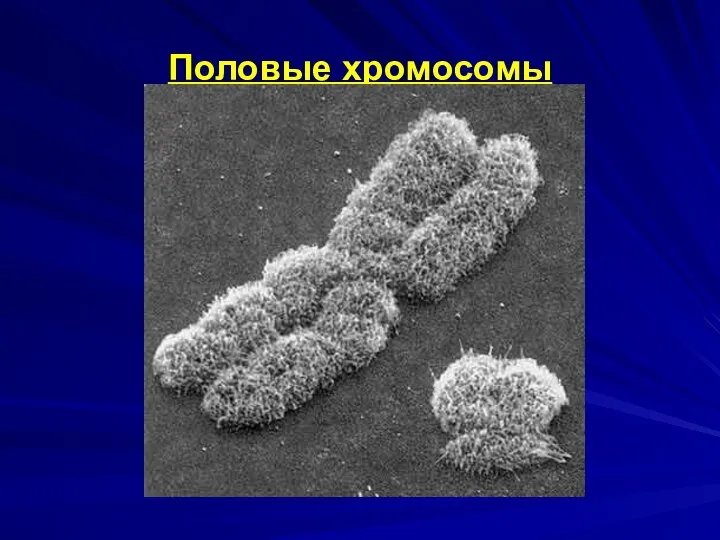
Половые хромосомы
Половые хромосомы

Х- и Y-хромосомы совершенно разные, кроме псевдоаутосомных участков.
Псевдоаутосомные участки:
располагаются на концах
Х- и Y-хромосомы совершенно разные, кроме псевдоаутосомных участков.
Псевдоаутосомные участки:
располагаются на концах

Х-хромосома
Одна из самых больших, 5% от общей длины генома (155 миллионов химических
Х-хромосома
Одна из самых больших, 5% от общей длины генома (155 миллионов химических

инактивация – дозовая компенсация, происходит в эмбриональном периоде,
центр инактивации – Xic
инактивация – дозовая компенсация, происходит в эмбриональном периоде,
центр инактивации – Xic

Y-хромосома
Более 50 млн п.н., 6 млн – палиндромы.
Содержит 78 генов (большинство
Y-хромосома
Более 50 млн п.н., 6 млн – палиндромы.
Содержит 78 генов (большинство

Имеет место «перетасовка» генов, что:
способствует самовосстановлению и элиминации мутантных генов,
приводит к
способствует самовосстановлению и элиминации мутантных генов,
приводит к
Генетический материал стабилен, но возможны перестройки
Генетический материал стабилен, но возможны перестройки

Основа патологии – изменчивость
Основа патологии – изменчивость
Каждая форма изменчивости может привести к развитию болезни
Каждая форма изменчивости может привести к развитию болезни

Мутации могут:
элиминироваться из генофонда популяции (т.к. клетки и организмы оказываются нежизнеспособными,
Мутации могут:
элиминироваться из генофонда популяции (т.к. клетки и организмы оказываются нежизнеспособными,

Хромосомные мутации
Изменения числа хромосом
Гаплоидия
Полиплоидия
Гетероплоидия
Трисомия
Моносомия
Нулисомия
Изменения структуры хромосом
Делеция
Дупликация
Инверсия
Транслокация
Инсерция
Хромосомные мутации
Изменения числа хромосом
Гаплоидия
Полиплоидия
Гетероплоидия
Трисомия
Моносомия
Нулисомия
Изменения структуры хромосом
Делеция
Дупликация
Инверсия
Транслокация
Инсерция

Трисомии по 8 и 22 парам
Трисомии по 8 и 22 парам

Моносомия 45,Х
Моносомия 45,Х

Делеция 22р-
Делеция 22р-

Дупликация
Дупликация
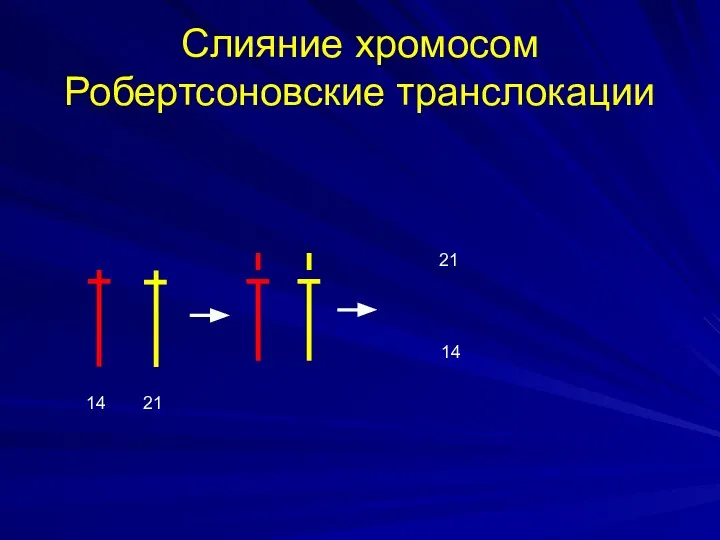
Слияние хромосом Робертсоновские транслокации
14
21
14
21
Слияние хромосом Робертсоновские транслокации
14
21
14
21
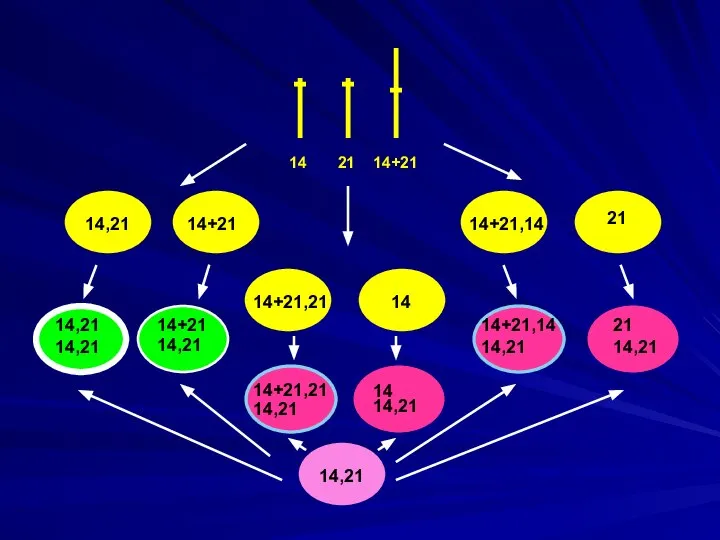
14 21 14+21
14,21
14+21
21
14+21,14
14+21,21
14
14,21
14,21
14,21
14+21
14,21
14+21,21
14,21
14
14,21
14+21,14
14,21
21
14,21
14 21 14+21
14,21
14+21
21
14+21,14
14+21,21
14
14,21
14,21
14,21
14+21
14,21
14+21,21
14,21
14
14,21
14+21,14
14,21
21
14,21
Кольцевая хромосома (17)
Кольцевая хромосома (17)

Изохромосомы
p
p p
q
q
q
q
p
p
p q
p
q
q
p
q
Изохромосомы
p
p p
q
q
q
q
p
p
p q
p
q
q
p
q
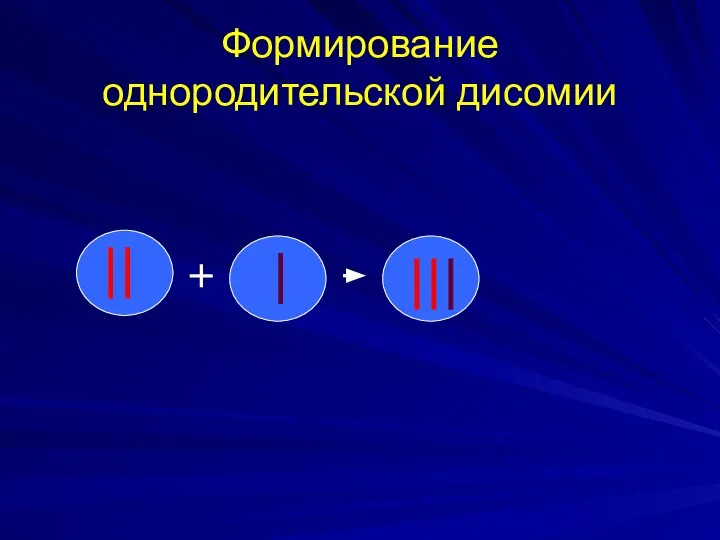
Формирование однородительской дисомии
Формирование однородительской дисомии

Наследственность и патология
Причина наследственной патологии – наследственная изменчивость
Мутационный процесс приводит к:
социальной
Наследственность и патология
Причина наследственной патологии – наследственная изменчивость
Мутационный процесс приводит к:
социальной

Классификация болезней
Наследственные болезни
Болезни с наследственной предрасположенностью (бÓльшее значение имеет генетический материал)
Болезни
Классификация болезней
Наследственные болезни
Болезни с наследственной предрасположенностью (бÓльшее значение имеет генетический материал)
Болезни

Наследственные болезни
Теоретически число наследственных болезней может составлять 50 000 – 100
Наследственные болезни
Теоретически число наследственных болезней может составлять 50 000 – 100

Классификация
наследственных болезней
Генетическая
Клиническая
Патогенетическая
Классификация
наследственных болезней
Генетическая
Клиническая
Патогенетическая

Генетическая классификация
хромосомные болезни,
генные болезни,
болезни с предрасположением (моногенные и полигенные),
генетические болезни соматических
Генетическая классификация
хромосомные болезни,
генные болезни,
болезни с предрасположением (моногенные и полигенные),
генетические болезни соматических

Выделяют также:
митохондриальные болезни,
болезни импринтинга,
эпигенетические болезни,
болезни экспансии тринуклеотидных повторов,
прионные болезни.
митохондриальные болезни,
болезни импринтинга,
эпигенетические болезни,
болезни экспансии тринуклеотидных повторов,
прионные болезни.

Хромосомные болезни
Причина – хромосомные мутации.
Типы генетических эффектов при хромосомных нарушениях:
специфические,
полуспецифические,
неспецифические.
Хромосомные болезни
Причина – хромосомные мутации.
Типы генетических эффектов при хромосомных нарушениях:
специфические,
полуспецифические,
неспецифические.


Фенотипическое проявление хромосомных мутаций зависит от:
типа мутации,
типа хромосомы,
типа поврежденного сегмента
Фенотипическое проявление хромосомных мутаций зависит от:
типа мутации,
типа хромосомы,
типа поврежденного сегмента

Классификация хромосомных болезней
Болезни, связанные с изменением числа хромосом.
Болезни, обусловленные изменением структуры
Классификация хромосомных болезней
Болезни, связанные с изменением числа хромосом.
Болезни, обусловленные изменением структуры

При классификации хромосомных болезней необходимо учитывать следующие моменты:
а) характеристика мутации
При классификации хромосомных болезней необходимо учитывать следующие моменты:
а) характеристика мутации

Для точной диагностики хромосомных болезней определяют:
тип мутации,
мутантную хромосому,
форму болезни (полная
Для точной диагностики хромосомных болезней определяют:
тип мутации,
мутантную хромосому,
форму болезни (полная

Синдром Дауна
(47,XX+21 или 47,XY+21)
Кариотип при синдроме Дауна
Синдром Дауна
(47,XX+21 или 47,XY+21)
Кариотип при синдроме Дауна

Частота 1:750 новорождённых.
Характерна малая средняя продолжительность жизни (35 лет).
Цитогенетические
Частота 1:750 новорождённых.
Характерна малая средняя продолжительность жизни (35 лет).
Цитогенетические
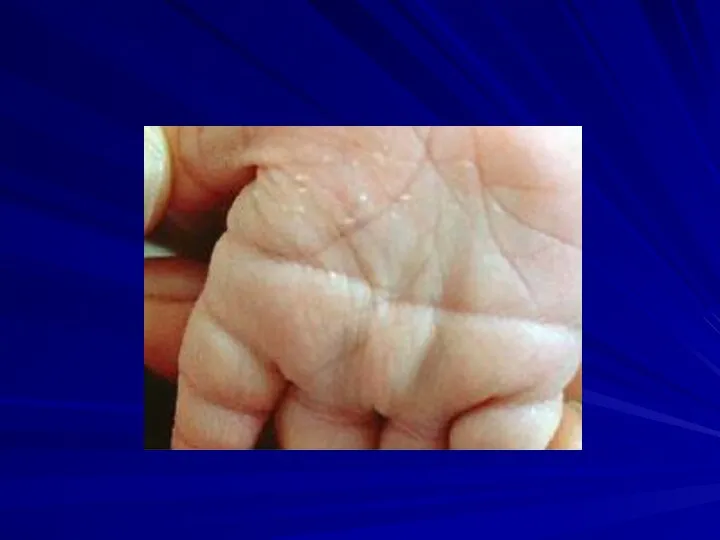



Синдром Патау
(47,XX+13 или 47,XY+13)
Кариотип при синдроме Патау
Синдром Патау
(47,XX+13 или 47,XY+13)
Кариотип при синдроме Патау

Частота – 1:6000
Соотношение полов 1:1
Цитогенетические варианты:
полная трисомия,
мозаицизм,
робертсоновская транслокация,
изохромосомы.
Частота – 1:6000
Соотношение полов 1:1
Цитогенетические варианты:
полная трисомия,
мозаицизм,
робертсоновская транслокация,
изохромосомы.

Фенотипические проявления:
сниженная масса тела,
микроцефалия,
недоразвитие мозга,
аномалии лица (запавшая
Фенотипические проявления:
сниженная масса тела,
микроцефалия,
недоразвитие мозга,
аномалии лица (запавшая
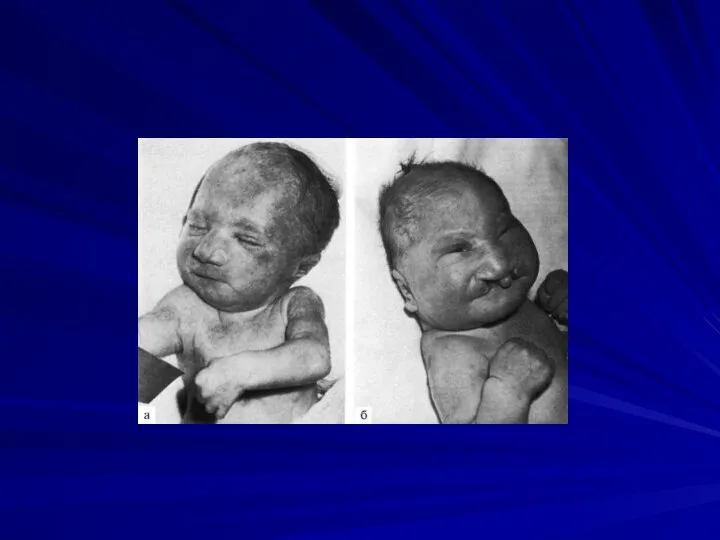

Синдром Эдвардса
(47,XX+18 или 47,XY+18)
Кариотип при синдроме Эдвардса
Синдром Эдвардса
(47,XX+18 или 47,XY+18)
Кариотип при синдроме Эдвардса

Частота 1:5000 – 1:7000
Соотношение частоты заболевания у девочек и мальчиков
Частота 1:5000 – 1:7000
Соотношение частоты заболевания у девочек и мальчиков

Фенотипические проявления:
Врожденные пороки развития:
лицевого черепа,
сердца (дефект межжелудочковой перегородки и
Фенотипические проявления:
Врожденные пороки развития:
лицевого черепа,
сердца (дефект межжелудочковой перегородки и
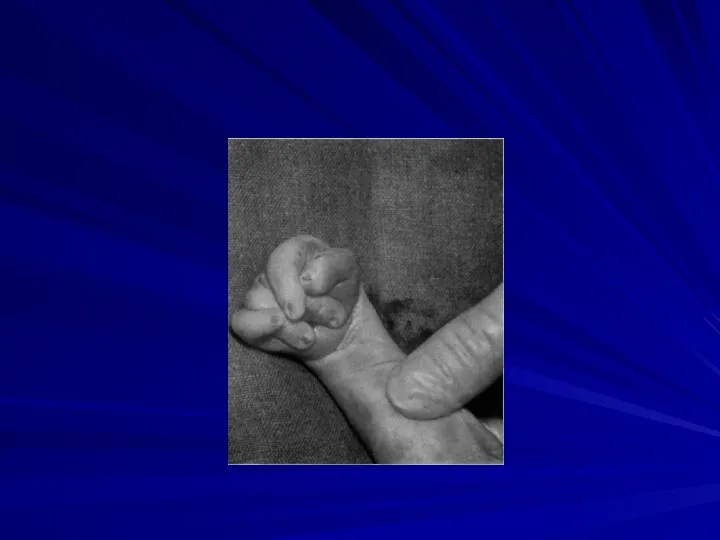

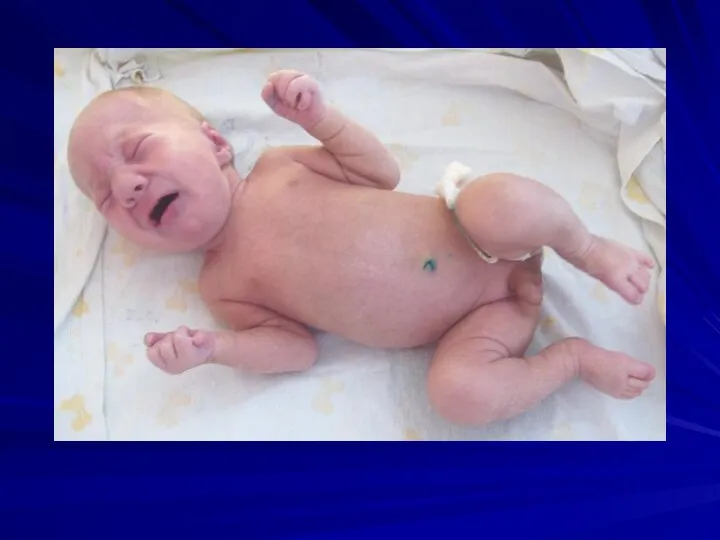
Синдром Шерешевского-Тернера
(45,X)
Частота 1:2000 – 1:5000 девочек
Цитогенетические варианты:
полная моносомия 45,Х (50%
Синдром Шерешевского-Тернера
(45,X)
Частота 1:2000 – 1:5000 девочек
Цитогенетические варианты:
полная моносомия 45,Х (50%

Основные клинические признаки:
нанизм,
крыловидные кожные складки на шее,
короткая шея
Основные клинические признаки:
нанизм,
крыловидные кожные складки на шее,
короткая шея


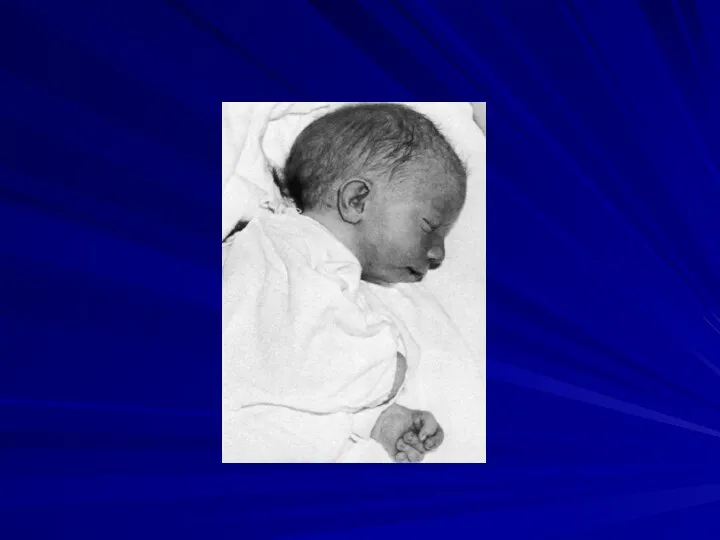
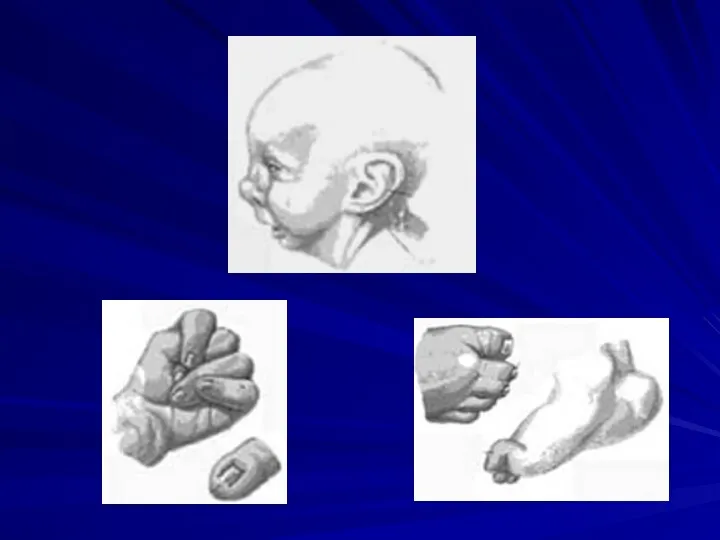
Синдром трисомии 8
(47,XX+8 или 47,XY+8)
Клиническая картина синдрома впервые описана разными авторами
Синдром трисомии 8
(47,XX+8 или 47,XY+8)
Клиническая картина синдрома впервые описана разными авторами

Основные клинические признаки:
отставание в умственном развитии,
отклонения в строении лица (выступающий
Основные клинические признаки:
отставание в умственном развитии,
отклонения в строении лица (выступающий

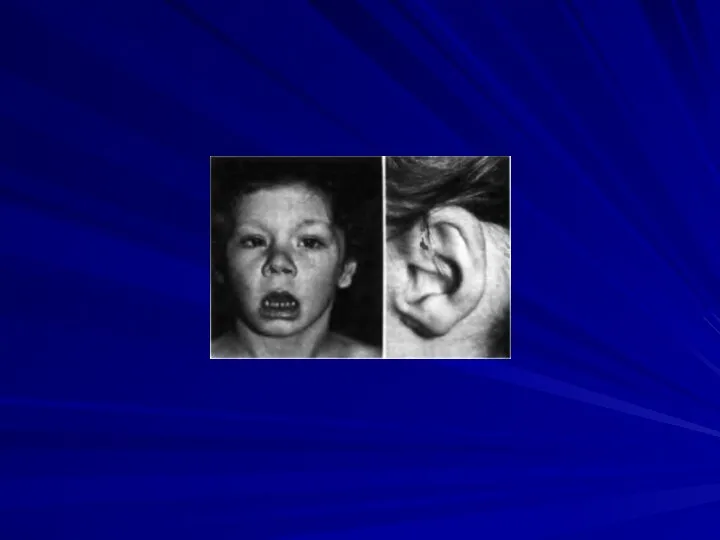


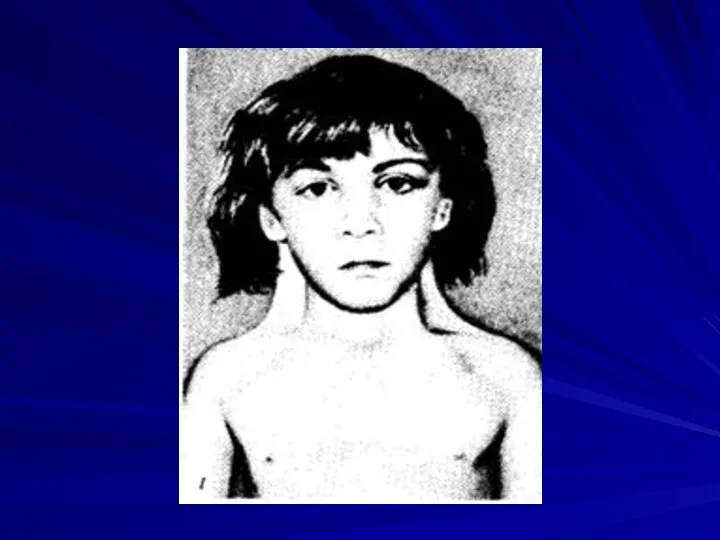
Синдром трисомии 14
(47,XX+14 или 47,XY+14)
Описан в 1975 году.
Прогноз жизни
Синдром трисомии 14
(47,XX+14 или 47,XY+14)
Описан в 1975 году.
Прогноз жизни

Основные клинические признаки:
микроцефалия,
асимметрия лица,
высокий и выступающий лоб,
нос
Основные клинические признаки:
микроцефалия,
асимметрия лица,
высокий и выступающий лоб,
нос

Синдроме Клайнфельтера (47,XXY, 48,XXXY и т.д.)
Кариотип при синдроме Клайнфельтера
Синдроме Клайнфельтера (47,XXY, 48,XXXY и т.д.)
Кариотип при синдроме Клайнфельтера

Частота 1:1000 мальчиков (1:500 – 1:750)
Цитогенетические варианты:
80% - ХХY, 20%
Частота 1:1000 мальчиков (1:500 – 1:750)
Цитогенетические варианты:
80% - ХХY, 20%

Синдром Лежена - синдром кошачьего крика
(46,XX5p- или 46,XY5p-)
Кариотип при синдроме Лежена
Синдром Лежена - синдром кошачьего крика
(46,XX5p- или 46,XY5p-)
Кариотип при синдроме Лежена

Частота 1:45000 новорожденных
Цитогенетические варианты:
образование кольцевой хромосомы 5 с частичной потерей
Частота 1:45000 новорожденных
Цитогенетические варианты:
образование кольцевой хромосомы 5 с частичной потерей

Основные клинические признаки:
нарушения гортани и специфический плач,
микроцефалия,
умственная отсталость,
Основные клинические признаки:
нарушения гортани и специфический плач,
микроцефалия,
умственная отсталость,
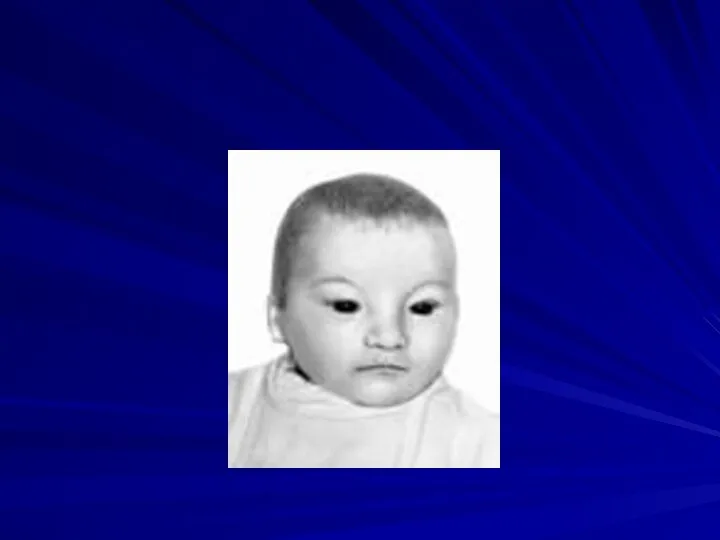
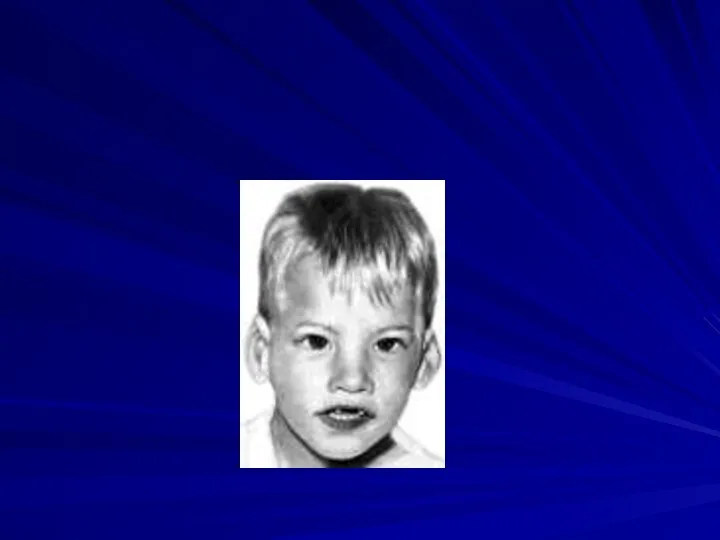

Синдром Вольфа-Хиршхорна
(46,XX4p- или 46,XY4p-)
Частота 1:100 000
Жизнеспособность детей резко снижена, большинство
Синдром Вольфа-Хиршхорна
(46,XX4p- или 46,XY4p-)
Частота 1:100 000
Жизнеспособность детей резко снижена, большинство

Основные клинические признаки:
более чем 50% детей имеют пороки внутренних органов
Основные клинические признаки:
более чем 50% детей имеют пороки внутренних органов
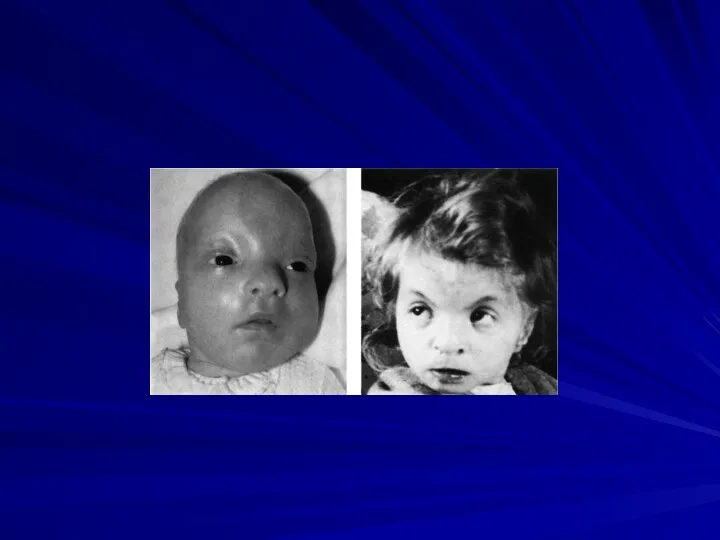

Ретинобластома (микроцитогенетический синдром 13q-14)
(46,XX13q- или 46,XY13q-)
Частота 1:15 000 – 1:34
Ретинобластома (микроцитогенетический синдром 13q-14)
(46,XX13q- или 46,XY13q-)
Частота 1:15 000 – 1:34
 Drugs. Addictions
Drugs. Addictions Влияние экстрагенитальной патологии на становление менструальной функции у девочек-подростков
Влияние экстрагенитальной патологии на становление менструальной функции у девочек-подростков Оптичні ілюзії. Види оптичних ілюзій
Оптичні ілюзії. Види оптичних ілюзій Роговица глаза. Аномалии развития роговицы
Роговица глаза. Аномалии развития роговицы Эндовидеохир. Разница между эндоскопической хирургией и классическим хирургическим вмешательством
Эндовидеохир. Разница между эндоскопической хирургией и классическим хирургическим вмешательством Препараты. Карнитина хлорид
Препараты. Карнитина хлорид Рахит. Спазмофилия. Гипервитаминоз D
Рахит. Спазмофилия. Гипервитаминоз D Медицинская реабилитация при сколиозе
Медицинская реабилитация при сколиозе Общее понятие о массаже и его физиологическом воздействии на организм
Общее понятие о массаже и его физиологическом воздействии на организм Острые нарушения кровообращения. Диагностика, неотложная помощь
Острые нарушения кровообращения. Диагностика, неотложная помощь Влияние экстрагенитальной патологии на становление менструальной функции у девочек-подростков
Влияние экстрагенитальной патологии на становление менструальной функции у девочек-подростков Носовые кровотечения
Носовые кровотечения Волчья пасть
Волчья пасть ВИЧ-инфекция. Статистика внутрибольничной заболеваемости
ВИЧ-инфекция. Статистика внутрибольничной заболеваемости Психологічна реабілітація. Загальне уявлення про реабілітацію. Тема 1
Психологічна реабілітація. Загальне уявлення про реабілітацію. Тема 1 Балалардың жедел ішек инфекциялары
Балалардың жедел ішек инфекциялары Составление алгоритмов оказания неотложной доврачебной помощи
Составление алгоритмов оказания неотложной доврачебной помощи Конфликт. Что делать
Конфликт. Что делать Жіті іш синдромы
Жіті іш синдромы Психология негіздері және коммуникативтік дағдылар кафедрасы
Психология негіздері және коммуникативтік дағдылар кафедрасы Методики определения предпочтительного типа будущей профессии
Методики определения предпочтительного типа будущей профессии Этическое и правовое регулирование биоэтических ситуаций. (Семинар 7. Тема 13.2)
Этическое и правовое регулирование биоэтических ситуаций. (Семинар 7. Тема 13.2) Причины и механизмы развитие иммунодефицитных состояний у сельскохозяйственных животных
Причины и механизмы развитие иммунодефицитных состояний у сельскохозяйственных животных Объекты и методы гистологических исследований. Основы цитологии
Объекты и методы гистологических исследований. Основы цитологии Геморрагический шок – диагностика и тактика поведения среднего медперсонала
Геморрагический шок – диагностика и тактика поведения среднего медперсонала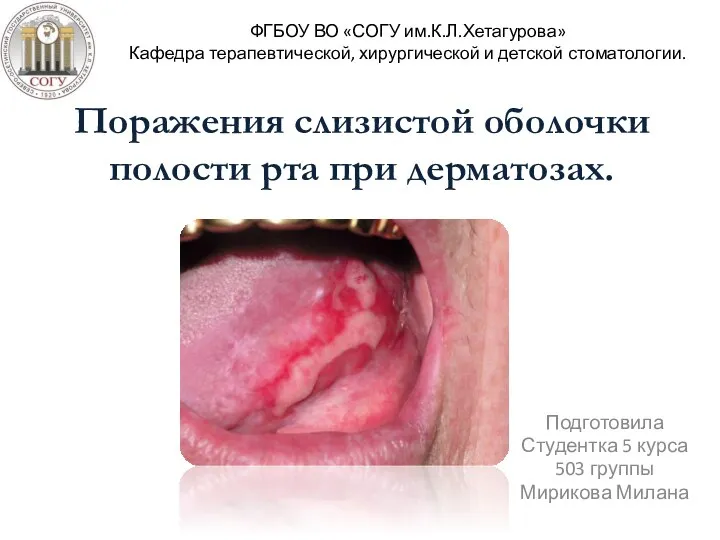 Поражения слизистой оболочки полости рта при дерматозах
Поражения слизистой оболочки полости рта при дерматозах The digestive system
The digestive system 26 апреля 2021 года — 35-я годовщина Чернобыльской аварии
26 апреля 2021 года — 35-я годовщина Чернобыльской аварии