Мітохондріальні хвороби. Загальна характеристика мітохондріальної патології. Клініка, діагностика, лікування
- Мітохондріальні хвороби. Загальна характеристика мітохондріальної патології. Клініка, діагностика, лікування

Содержание
- 2. План 1.Характеристика мітохондріального геному. 2.Етіопатогенез мітохондріальних захворювань. 3.Класифікація мітохондропатій. 4.Клініка найбільш поширених мітохондропатій. 5.Принципи лікування мітохондропатій.
- 3. Актуальність теми Серед сучасних спадкових хвороб, які зустрічаються у людей, існує безліч нозологічних одиниць, які повністю
- 4. Актуальність теми Мітохондропатії займають високу питому вагу серед здорового населення 1,6%, а серед хворих із множинним
- 5. Мітохондії — одні з найбільших органел клітини, які містяться в усіх евкаріотичних клітинах, окрім еритроцитів і
- 6. Структура мітохондрій Мітохондрії мають свої особисті ДНК, РНК і рибосоми, самі синтезують частину своїх білків, розмножуються
- 7. Кожна клітина людського організму містить сотні мітохондрій і тисячі ДНК мітохондрій. Якщо мутація виникає в одній
- 8. Особливості мітохондріальної ДНК: Строго материнський характер успадкування ДНК, тобто вони передаються від матері до дочок та
- 9. Мітохондріальні захворювання - одна з найбільших класів дегенеративних хвороб нервово-м'язової системи, що має тяжкий проградієнтний перебіг,
- 10. Класифікація мітохондропатій Мітохондріальні хвороби класифікують за типом мутацій: 1 .Місенс – мутантні мітохондріальні хвороби: нейроофтальмопатія Лебера;
- 11. Класифікація мітохондропатій 3. Мітохондріальні хвороби, які викликані делеціями або дуплікаціями мітохондріальної ДНК: синдром Кернса-Сейра; синдром Пірсона;
- 12. Класифікація мітохондропатій 4. Хвороби, які викликані мутаціями, що знижують число копій мітохондріальної ДНК: летальна інфантильна дихальна
- 13. Загальні клінічні риси МТХЗ Полісиндромність уражень з частим залученням нервової системи органа зору, серця і соматичних
- 14. Сутність молекулярно-генетичних досліджень при мітохондропатіях. Мутації мітохондріальної ДНК визначаються у зразках м′язової тканини методом полімеразної ланцюгової
- 15. Синдром Лебера (спадкова атрофія зорових нервів) уперше описаний у 1971р. Теодором Лебером. Класична назва синдрому —
- 16. Синдром Лебера (спадкова атрофія зорових нервів)
- 17. Синдром Лебера (спадкова атрофія зорових нервів) Критерії діагнозу: Материнський тип успадкування; Характерні клінічні ознаки; Ідентифікація за
- 18. Синдром Лебера (спадкова атрофія зорових нервів) Диференціальну діагностику потрібно проводити із захворюваннями, що супроводжуються, зниженням гостроти
- 19. Мутації в генах тРНК Синдром MERRF (міоклонус-епілепсія, рвані червоні волокна", mioclonus-epilesy, rend red fibre ). Уперше
- 20. Вік хворого на початку захворювання — від 3 до 65 років. Ранніми клінічними ознаками є швидка
- 21. Критерії діагнозу: материнський тип успадкування; початок захворювання у віці 3—65 років; ЦНС — міоклонус, атаксія, деменція
- 22. Диференціальну діагностику проводять з іншими прогресивними міокло- нус-епілепсіями ( хворобою Ґоше, синдромом міоклонуса з нирковою недостатністю
- 23. Синдром MELAS (мітохондріальна енцефалопатія, лактат-ацидоз, інсуль-топодібні епізоди). Уперше його виділив у нозологічно самостійну форму S.G. Pavlakis
- 24. Основні діагностичні критерії: непереносимість фізичних навантажень. початок захворювання до 40 років (частіше до 20 років). інсультоподібні
- 25. Додаткові діагностичні симптоми: кальцифікація базальних гангліїв при комп‘ютерній томографії (КТ) або магнітно – резонансній томографії (МРТ)
- 26. Клінічна картина: Перші ознаки частіше з‘являються у віці 6–10 років, хоча можливі як більш ранній початок
- 27. Клінічна картина: Інсультоподібні епізоди проявляються рецидивуючими нападами головного болю, запамороченням, розвитком вогнищевої неврологічної симптоматики (парези, паралічі
- 28. Критерії діагнозу: материнський тип успадкування; вік маніфестації — до 40 років; мігренеподібний головний біль із нудотою
- 29. Делеції або дуплікації ділянок мітохондріальної ДНК Синдром Кернса—Сейра вперше описано в 1958 р. під назвою "пігментний
- 30. Основні діагностичні критерії: Вік хворого при маніфестації хвороби - 4-18 років. Прогресуюча зовнішня офтальмоплегія. Пігментний ретиніт
- 31. Клінічні прояви: М'язова система: птоз, як правило, симетричний і білатеральний; повільно прогресуюча зовнішня офтальмоплегія; низхідний характер
- 32. Клінічні прояви: Орган зору: пігментний ретиніт; на очному дні пігментна грануляція типу "перець з сіллю"; атрофія
- 33. Клінічні прояви: Ендокринна система: низькорослість; гіпогонадизм; гінекомастія; цукровий діабет; гіперальдостеронізм; гіпопаратиреоз. Факультативні ознаки: скелетні аномалії (кіфосколіоз,
- 34. Критерії діагнозу: початок захворювання — у віці 4—18 років; мозочковий синдром з інтенційним тремором; зниження інтелекту;
- 35. Диференціальна діагностика інші форми прогресивних міопатій, а також із захворюваннями, що поєднуються з птозом (міастенія, діабетична
- 36. Синдром Пірсона Уперше описав у 1979 p. H.A. Pearson із співавт. під назвою "рефрактерна сідеробластна анемія
- 37. Критерії діагнозу: Критерії діагнозу: початок захворювання — від народження або в перші місяці життя; гіпопластична анемія;
- 38. Синдроми множинних делецїй мітохондріальних ДНК Уперше множинні делеції виявлено у хворих з автосомно-домінантною прогресивною зовнішньою офтальмоплегією,
- 39. Синдроми множинних делецїй мітохондріальних ДНК Серед кардинальних симптомів хвороби виділяють очні (прогресивний птоз, зовнішня офтальмоплегія, офтальмопарез)
- 40. Критерії діагнозу: блефароптоз, зовнішня офтальмоплегія; м'язова слабкість; ЦНС — нейросенсорна глухота, атрофія зорових нервів; прогресивний перебіг;
- 41. Делеція мітохондріальної ДНК Уперше делеції мтДНК описали С.Т. Moraes із співавт. у 1991 р. Захворювання успадковується
- 42. Критерії діагнозу:
- 43. Мутації в ядерній ДНК. Глутарова ацидемія хвороба зумовлена множинним дефіцитом мітохондріальних флавопротеїнових ацил-КоА-дегідрогеназ. Тип успадковування глутарової
- 44. Критерії діагнозу глутарової ацидемії. Перша форма: респіраторний дистрес-синдром; м'язова гіпотонія; блювання; гепатоспленомегалія; незвичний запах сечі; анемія;
- 45. Критерії діагнозу глутарової ацидемії. Друга форма: рання маніфестація; важкий перебіг; рання смерть. Третя форма: початок на
- 46. Критерії діагнозу глутарової ацидемії Четверта форма: шлунково-кишкові розлади; гепатомегалія; жовтяниця; м’язова слабість.
- 47. Фумарова ацидемія. Вперше описаний Zinn і соавт. в 1968 році. Локус - lq42.
- 48. Критерії діагнозу фумарової ацидемії. тип успадкування - аутосомно-рецесивнии; маніфестація до 5-7 місяців; погана прибавка маси; повторні
- 49. Критерії діагнозу фумарової ацидемії. ензімодіагностика - дефект фумарази; в сечі: висока концентрація фумарової кислоти; - в
- 50. Принципи лікування мітохондропатій 1.Дієтичні заходи. 2. Додаткове введення кофакторів, які беруть участь в ензимних реакціях енергетичного
- 52. Скачать презентацию

План
1.Характеристика мітохондріального геному.
2.Етіопатогенез мітохондріальних захворювань.
3.Класифікація мітохондропатій.
4.Клініка найбільш поширених мітохондропатій.
5.Принципи лікування мітохондропатій.
План
1.Характеристика мітохондріального геному.
2.Етіопатогенез мітохондріальних захворювань.
3.Класифікація мітохондропатій.
4.Клініка найбільш поширених мітохондропатій.
5.Принципи лікування мітохондропатій.

Актуальність теми
Серед сучасних спадкових хвороб, які зустрічаються у людей, існує безліч
Актуальність теми
Серед сучасних спадкових хвороб, які зустрічаються у людей, існує безліч

Актуальність теми
Мітохондропатії займають високу питому вагу серед здорового населення 1,6%, а
Актуальність теми
Мітохондропатії займають високу питому вагу серед здорового населення 1,6%, а

Мітохондії — одні з найбільших органел клітини, які містяться в усіх
Мітохондії — одні з найбільших органел клітини, які містяться в усіх
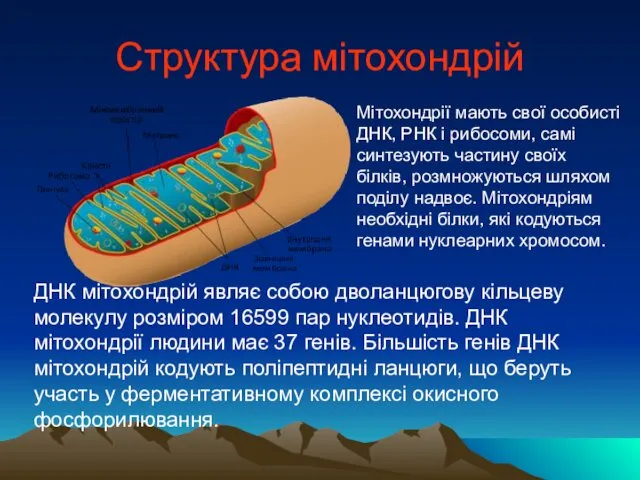
Структура мітохондрій
Мітохондрії мають свої особисті ДНК, РНК і рибосоми, самі синтезують
Структура мітохондрій
Мітохондрії мають свої особисті ДНК, РНК і рибосоми, самі синтезують

Кожна клітина людського організму містить сотні мітохондрій і тисячі ДНК мітохондрій.
Кожна клітина людського організму містить сотні мітохондрій і тисячі ДНК мітохондрій.

Особливості мітохондріальної ДНК:
Строго материнський характер успадкування ДНК, тобто вони передаються від
Особливості мітохондріальної ДНК:
Строго материнський характер успадкування ДНК, тобто вони передаються від

Мітохондріальні захворювання - одна з найбільших класів дегенеративних хвороб нервово-м'язової системи,
Мітохондріальні захворювання - одна з найбільших класів дегенеративних хвороб нервово-м'язової системи,

Класифікація мітохондропатій
Мітохондріальні хвороби класифікують за типом мутацій:
1 .Місенс
Класифікація мітохондропатій
Мітохондріальні хвороби класифікують за типом мутацій:
1 .Місенс

Класифікація мітохондропатій
3. Мітохондріальні хвороби, які викликані делеціями або дуплікаціями мітохондріальної ДНК:
синдром Кернса-Сейра;
синдром
Класифікація мітохондропатій
3. Мітохондріальні хвороби, які викликані делеціями або дуплікаціями мітохондріальної ДНК:
синдром Кернса-Сейра;
синдром

Класифікація мітохондропатій
4. Хвороби, які викликані мутаціями, що знижують число копій мітохондріальної
Класифікація мітохондропатій
4. Хвороби, які викликані мутаціями, що знижують число копій мітохондріальної

Загальні клінічні риси МТХЗ
Полісиндромність уражень з частим залученням нервової системи
Загальні клінічні риси МТХЗ
Полісиндромність уражень з частим залученням нервової системи

Сутність молекулярно-генетичних досліджень при мітохондропатіях.
Мутації мітохондріальної ДНК визначаються у зразках м′язової
Сутність молекулярно-генетичних досліджень при мітохондропатіях.
Мутації мітохондріальної ДНК визначаються у зразках м′язової

Синдром Лебера (спадкова атрофія зорових нервів)
уперше описаний у 1971р. Теодором
Синдром Лебера (спадкова атрофія зорових нервів)
уперше описаний у 1971р. Теодором
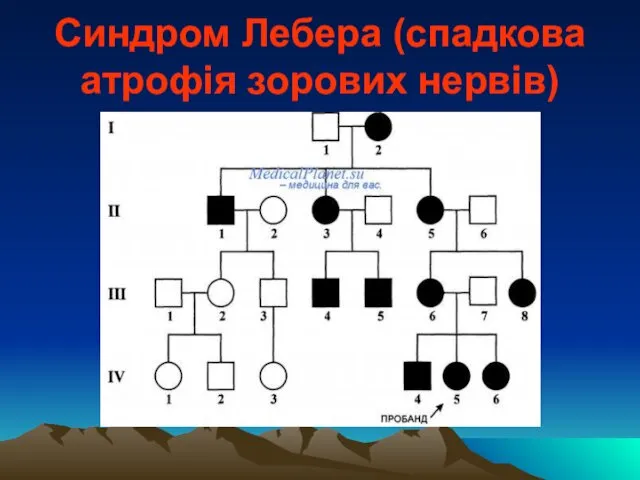
Синдром Лебера (спадкова атрофія зорових нервів)
Синдром Лебера (спадкова атрофія зорових нервів)

Синдром Лебера (спадкова атрофія зорових нервів)
Критерії діагнозу:
Материнський тип успадкування;
Характерні клінічні ознаки;
Ідентифікація
Синдром Лебера (спадкова атрофія зорових нервів)
Критерії діагнозу:
Материнський тип успадкування;
Характерні клінічні ознаки;
Ідентифікація

Синдром Лебера (спадкова атрофія зорових нервів)
Диференціальну діагностику потрібно проводити із захворюваннями,
Синдром Лебера (спадкова атрофія зорових нервів)
Диференціальну діагностику потрібно проводити із захворюваннями,

Мутації в генах тРНК
Синдром MERRF (міоклонус-епілепсія, рвані червоні волокна", mioclonus-epilesy,
Мутації в генах тРНК Синдром MERRF (міоклонус-епілепсія, рвані червоні волокна", mioclonus-epilesy,

Вік хворого на початку захворювання — від 3 до 65 років.
Вік хворого на початку захворювання — від 3 до 65 років.

Критерії діагнозу:
материнський тип успадкування;
початок захворювання у віці 3—65 років;
ЦНС — міоклонус,
Критерії діагнозу:
материнський тип успадкування;
початок захворювання у віці 3—65 років;
ЦНС — міоклонус,

Диференціальну діагностику проводять з іншими прогресивними міокло-
нус-епілепсіями ( хворобою Ґоше, синдромом
Диференціальну діагностику проводять з іншими прогресивними міокло-
нус-епілепсіями ( хворобою Ґоше, синдромом

Синдром MELAS (мітохондріальна енцефалопатія, лактат-ацидоз, інсуль-топодібні епізоди).
Уперше його виділив у
Синдром MELAS (мітохондріальна енцефалопатія, лактат-ацидоз, інсуль-топодібні епізоди).
Уперше його виділив у

Основні діагностичні критерії:
непереносимість фізичних навантажень.
початок захворювання до 40 років
Основні діагностичні критерії:
непереносимість фізичних навантажень.
початок захворювання до 40 років

Додаткові діагностичні симптоми:
кальцифікація базальних гангліїв при комп‘ютерній томографії (КТ) або
Додаткові діагностичні симптоми:
кальцифікація базальних гангліїв при комп‘ютерній томографії (КТ) або

Клінічна картина:
Перші ознаки частіше з‘являються у віці 6–10 років, хоча
Клінічна картина:
Перші ознаки частіше з‘являються у віці 6–10 років, хоча

Клінічна картина:
Інсультоподібні епізоди проявляються рецидивуючими нападами головного болю, запамороченням, розвитком вогнищевої
Клінічна картина:
Інсультоподібні епізоди проявляються рецидивуючими нападами головного болю, запамороченням, розвитком вогнищевої

Критерії діагнозу:
материнський тип успадкування;
вік маніфестації — до 40 років;
мігренеподібний головний біль
Критерії діагнозу:
материнський тип успадкування;
вік маніфестації — до 40 років;
мігренеподібний головний біль

Делеції або дуплікації ділянок мітохондріальної ДНК
Синдром Кернса—Сейра
вперше описано в 1958
Делеції або дуплікації ділянок мітохондріальної ДНК
Синдром Кернса—Сейра
вперше описано в 1958

Основні діагностичні критерії:
Вік хворого при маніфестації хвороби - 4-18 років.
Прогресуюча зовнішня
Основні діагностичні критерії:
Вік хворого при маніфестації хвороби - 4-18 років.
Прогресуюча зовнішня

Клінічні прояви:
М'язова система:
птоз, як правило, симетричний і білатеральний;
повільно прогресуюча зовнішня офтальмоплегія;
низхідний
Клінічні прояви:
М'язова система:
птоз, як правило, симетричний і білатеральний;
повільно прогресуюча зовнішня офтальмоплегія;
низхідний

Клінічні прояви:
Орган зору:
пігментний ретиніт;
на очному дні пігментна грануляція типу "перець з
Клінічні прояви:
Орган зору:
пігментний ретиніт;
на очному дні пігментна грануляція типу "перець з

Клінічні прояви:
Ендокринна система:
низькорослість;
гіпогонадизм;
гінекомастія;
цукровий діабет;
гіперальдостеронізм;
гіпопаратиреоз.
Факультативні ознаки:
скелетні аномалії (кіфосколіоз, краніосиностоз, метафізарна дисплазія, гіпоплазія
Клінічні прояви:
Ендокринна система:
низькорослість;
гіпогонадизм;
гінекомастія;
цукровий діабет;
гіперальдостеронізм;
гіпопаратиреоз.
Факультативні ознаки:
скелетні аномалії (кіфосколіоз, краніосиностоз, метафізарна дисплазія, гіпоплазія

Критерії діагнозу:
початок захворювання — у віці 4—18 років;
мозочковий синдром з інтенційним
Критерії діагнозу:
початок захворювання — у віці 4—18 років;
мозочковий синдром з інтенційним

Диференціальна діагностика
інші форми прогресивних міопатій, а також із захворюваннями,
Диференціальна діагностика
інші форми прогресивних міопатій, а також із захворюваннями,

Синдром Пірсона
Уперше описав у 1979 p. H.A. Pearson із співавт.
Синдром Пірсона
Уперше описав у 1979 p. H.A. Pearson із співавт.

Критерії діагнозу:
Критерії діагнозу:
початок захворювання — від народження або в перші місяці
Критерії діагнозу:
Критерії діагнозу:
початок захворювання — від народження або в перші місяці

Синдроми множинних делецїй мітохондріальних ДНК
Уперше множинні делеції виявлено у хворих
Синдроми множинних делецїй мітохондріальних ДНК
Уперше множинні делеції виявлено у хворих

Синдроми множинних делецїй мітохондріальних ДНК
Серед кардинальних симптомів хвороби виділяють очні (прогресивний
Синдроми множинних делецїй мітохондріальних ДНК
Серед кардинальних симптомів хвороби виділяють очні (прогресивний

Критерії діагнозу:
блефароптоз, зовнішня офтальмоплегія;
м'язова слабкість;
ЦНС — нейросенсорна глухота, атрофія зорових нервів;
прогресивний
Критерії діагнозу:
блефароптоз, зовнішня офтальмоплегія;
м'язова слабкість;
ЦНС — нейросенсорна глухота, атрофія зорових нервів;
прогресивний

Делеція мітохондріальної ДНК
Уперше делеції мтДНК описали С.Т. Moraes із співавт. у
Делеція мітохондріальної ДНК
Уперше делеції мтДНК описали С.Т. Moraes із співавт. у

Критерії діагнозу:
Критерії діагнозу:

Мутації в ядерній ДНК.
Глутарова ацидемія
хвороба зумовлена множинним дефіцитом мітохондріальних флавопротеїнових
Мутації в ядерній ДНК.
Глутарова ацидемія
хвороба зумовлена множинним дефіцитом мітохондріальних флавопротеїнових

Критерії діагнозу глутарової ацидемії.
Перша форма:
респіраторний дистрес-синдром;
м'язова гіпотонія;
блювання;
гепатоспленомегалія;
незвичний запах сечі;
анемія;
лицеві дисморфії.
Критерії діагнозу глутарової ацидемії.
Перша форма:
респіраторний дистрес-синдром;
м'язова гіпотонія;
блювання;
гепатоспленомегалія;
незвичний запах сечі;
анемія;
лицеві дисморфії.

Критерії діагнозу глутарової ацидемії.
Друга форма:
рання маніфестація;
важкий перебіг;
рання смерть.
Третя форма:
початок на першому
Критерії діагнозу глутарової ацидемії.
Друга форма:
рання маніфестація;
важкий перебіг;
рання смерть.
Третя форма:
початок на першому

Критерії діагнозу глутарової ацидемії
Четверта форма:
шлунково-кишкові розлади;
гепатомегалія;
жовтяниця;
м’язова слабість.
Критерії діагнозу глутарової ацидемії
Четверта форма:
шлунково-кишкові розлади;
гепатомегалія;
жовтяниця;
м’язова слабість.

Фумарова ацидемія.
Вперше описаний Zinn і соавт. в 1968 році. Локус -
Фумарова ацидемія.
Вперше описаний Zinn і соавт. в 1968 році. Локус -

Критерії діагнозу фумарової ацидемії.
тип успадкування - аутосомно-рецесивнии;
маніфестація до 5-7 місяців;
погана прибавка
Критерії діагнозу фумарової ацидемії.
тип успадкування - аутосомно-рецесивнии;
маніфестація до 5-7 місяців;
погана прибавка

Критерії діагнозу фумарової ацидемії.
ензімодіагностика - дефект фумарази;
в сечі: висока концентрація фумарової
Критерії діагнозу фумарової ацидемії.
ензімодіагностика - дефект фумарази;
в сечі: висока концентрація фумарової

Принципи лікування мітохондропатій
1.Дієтичні заходи.
2. Додаткове введення кофакторів, які беруть участь в
Принципи лікування мітохондропатій
1.Дієтичні заходи.
2. Додаткове введення кофакторів, які беруть участь в
 Мій імідж як особистості та професіонала
Мій імідж як особистості та професіонала Пиелонефрит
Пиелонефрит Forme de comunicare
Forme de comunicare Дифференциальная диагностика при распространенных заболеваниях в терапии
Дифференциальная диагностика при распространенных заболеваниях в терапии Индивидуальное развитие детей школьного возраста
Индивидуальное развитие детей школьного возраста Презентация Анатомия
Презентация Анатомия Лечение гипертонического криза у пациентов с острой левожелудочковой недостаточностью (отеком легких)
Лечение гипертонического криза у пациентов с острой левожелудочковой недостаточностью (отеком легких) Гемостаз
Гемостаз Психологические причины возникновения опасных ситуаций . Ошибки принятия решения. (9)
Психологические причины возникновения опасных ситуаций . Ошибки принятия решения. (9) Преждевременные роды
Преждевременные роды Bhp masażysty
Bhp masażysty Вирус СПИД и человек – динамика борьбы
Вирус СПИД и человек – динамика борьбы Здоровый образ жизни - наш выбор
Здоровый образ жизни - наш выбор Сентимент талдау есебін шешу
Сентимент талдау есебін шешу Маститы. Анатомия и физиология молочной железы
Маститы. Анатомия и физиология молочной железы Фаст-фудтың химиялық құрамы мен адам ағзасына әсерін зерттеу
Фаст-фудтың химиялық құрамы мен адам ағзасына әсерін зерттеу Пищевые отравления
Пищевые отравления Неврозы
Неврозы Шум. Шумовое воздействие на организм
Шум. Шумовое воздействие на организм Основы интеллектуальной деятельности
Основы интеллектуальной деятельности Потребности новорожденного. Как вырабатывается молоко? Что нужно делать, чтобы было молоко. (Лекция 1)
Потребности новорожденного. Как вырабатывается молоко? Что нужно делать, чтобы было молоко. (Лекция 1) Паразитизм, патогенность и паразитарные системы
Паразитизм, патогенность и паразитарные системы Понятие аутизм и расстройство аутистического спектра
Понятие аутизм и расстройство аутистического спектра Острые пневмонии у детей
Острые пневмонии у детей Запалення у тварин. Стоматит у кішки. Гепатит С. Абсцес мозку
Запалення у тварин. Стоматит у кішки. Гепатит С. Абсцес мозку Диабеттік фетопатия
Диабеттік фетопатия Диссеминированные процессы в лёгких
Диссеминированные процессы в лёгких Тіл аурулары мен аномалиялары. Глоссалгия
Тіл аурулары мен аномалиялары. Глоссалгия