- Наследственные заболевания в практике педиатра и терапевта

Содержание
- 2. Генетика - наука о наследственности и изменчивости. Термин ввел в 1907 году У.Бэтсон. Он определил содержание
- 3. Генетика человека - фундаментальная наука Часть общей генетики, изучающей законы накопления, передачи и реализации информации о
- 4. Медицинская генетика - наука о роли наследственности в патологии человека, закономерностях передачи наследственных болезней, их диагностике,
- 5. Развитие генетики человека 1952 T.C. Hsu Открытие гипотонической обработки Дж. Уотсон, Ф. Крик 1953 г. Открытие
- 6. Развитие генетики человека Первый автоматический секвенатор, 1987 г. 1993 год Кэрри Мюллис Нобелевская премия в области
- 7. Развитие клинической генетики 1959 – Франсуа Лежен 47 хромосом (трисомия 21) при синдроме Дауна Джон Лэнгдон
- 8. Успехи клинической генетики в СССР С.И. Давиденков В 1925 г. классификация наследственных болезней должна быть "каталогом
- 9. В Москве в 1935 г был создан «Медико-генетический институт». Соломон Григорьевич Левит Были выполнены работы в
- 10. Развитие клинической генетики в СССР Постановлением Президиума АМН СССР 01.03.61 была создана Лаборатория медицинской генетики под
- 11. Основные нормативные документы, регулирующие вопросы редких заболеваний в РФ Федеральный закон РФ № 323-ФЗ «Об основах
- 12. Всемирная организация здравоохранения (ВОЗ) определяет РЗ следующим образом: 1. Редко встречаются в популяции населения (статистически редкое
- 13. Основные нормативные документы, регулирующие вопросы редких заболеваний в РФ Постановление Правительства РФ от 26 апреля 2012
- 14. ПЕРЕЧЕНЬ ЖИЗНЕУГРОЖАЮЩИХ И ХРОНИЧЕСКИХ ПРОГРЕССИРУЮЩИХ РЕДКИХ (ОРФАННЫХ) ЗАБОЛЕВАНИЙ, ПРИВОДЯЩИХ К СОКРАЩЕНИЮ ПРОДОЛЖИТЕЛЬНОСТИ ЖИЗНИ ГРАЖДАН ИЛИ ИХ
- 15. ПЕРЕЧЕНЬ ЖИЗНЕУГРОЖАЮЩИХ И ХРОНИЧЕСКИХ ПРОГРЕССИРУЮЩИХ РЕДКИХ (ОРФАННЫХ) ЗАБОЛЕВАНИЙ, ПРИВОДЯЩИХ К СОКРАЩЕНИЮ ПРОДОЛЖИТЕЛЬНОСТИ ЖИЗНИ ГРАЖДАН ИЛИ ИХ
- 16. Изменения в перечне 24-х нозологий В 2019 году программа «7 высоко-затратных нозологий» превратилась в программу «12
- 17. Из чего сделана ДНК – дезоксирибонуклеиновая кислота? 3,2 млрд. нуклеотидов → Человек Сахар (пентоза) Фосфат Азотистое
- 18. Код ДНК – четыре азотистых основания 3% ДНК – кодирующая часть генома (всей совокупности ДНК клетки).
- 19. Вторичная структура ДНК – двухцепочечная спираль A = T G ≡ C H2O CO2 CH4 O2
- 20. Третичная структура ДНК (суперспирализация ДНК) Хромосома – комплекс ДНК и белков 46 хромосом – 23 хромосомы
- 21. Методы анализа ДНК
- 22. Структурные элементы гена Экзон размер экзома человека — ~ 40 млн.н.п. Интрон Экзон-интронная граница Межгенные области
- 23. Распространенные виды мутаций Миссенс мутация Нонсенс мутация Молчащая мутация Мутация сдвига рамки считывания Мутация сайта сплайсинга
- 24. Методы анализа определения геномных вариантов ПДАФ (полиморфизм длин амплифицированных фрагментов) ПДРФ (полиморфизм длин рестрикционных фрагментов) АС-ПЦР
- 25. Виды геномных вариантов Мутация – патогенный геномный вариант Variant of unknown significance (вариант неизвестной значимости) Полиморфизм
- 26. Появление и развитие технологий секвенирования ДНК 1953 Расшифрована структура ДНК Д. Уотсон, Ф. Крик Applied Biosystems
- 27. Классификация наследственных болезней Хромосомные болезни, связанные с изменением их числа Хромосомные болезни, связанные с изменением структуры
- 28. Классификация наследственных болезней Генные болезни, связанные с точечными мутациями ядерной ДНК: Моногенные заболевания, связанные с мутациями
- 29. Классификация наследственных болезней Болезни, обусловленные дефектами митохондриальной ДНК: Генные, связанные с точечными мутациями мтДНК Болезни, обусловленные
- 30. Хромосомные болезни Числовые аномалии хромосом ~
- 31. Хромосомные болезни Структурные аномалии хромосом ~ 1 из 500 индивидов является носителем структурной аномалии Большинство сбалансированы
- 32. Структурные аномалии хромосом транслокации делеции дупликации инверсии инсерции кольцевые хромосомы маркерные хромосомы
- 33. Синдромы делеций протяженных последовательностей
- 34. Делеция 11p11.2p12 (синдром Потоки-Шейфер) OMIM# 601224 Отставание в умственном развитии Множественные экзостозы (EXT2 ) Теменные окна
- 37. Синдром Вильямса deletion 7q11.23q11.23 OMIM# 194050 Отставание в умственном развитии ВПС (75%) SVAS Гиперкальцемия Лицевые микроаномалии
- 40. Методы диагностики хромосомных заболеваний Клинические - выявление микроаномалий развития и пороков развития органов и систем Лабораторные
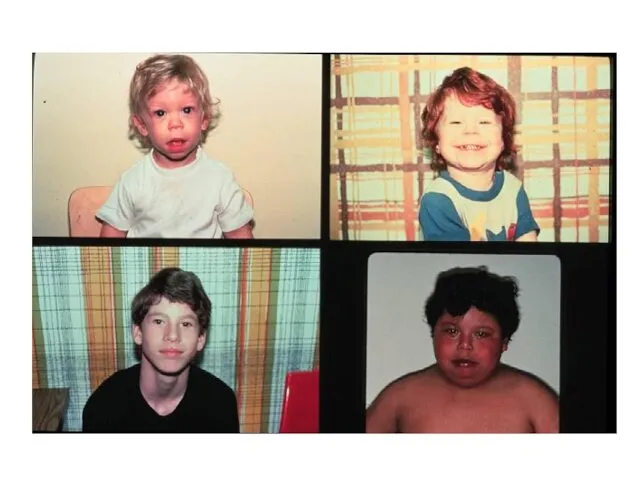
- 41. Клинические показания к применению цитогенетических и молекулярно- цитогенетических методов Краниофациальные микроаномалии: Долихоцефалия Круглое плоское лицо Широкие
- 42. Клинические показания к применению цитогенетических и молекулярно- цитогенетических методов Неспецифическая умственная отсталость в сочетании с микроаномалиями
- 43. Результаты лучевой диагностики Лиссэнцефалия (17.13.3 del, 22q11 del, Xp22 del, 2p25 del) Голопроэнцефалия (1q41-q42, 13q24-q34) Гипоплазия,
- 44. Генные болезни Связанные с точечными мутациями ядерной ДНК: моногенные заболевания, связанные с мутациями ядерной ДНК болезни
- 45. Типы наследования моногенных заболеваний Аутосомно-рецессивный (большинство заболеваний) Аутосомно-доминантный Х-сцепленный рецессивный Митохондриальный
- 46. Сложности клинического этапа диагностики моногенных заболеваний Наследственные заболевания редкие, представленные единичными наблюдениями. Широкий клинический полиморфизм. От
- 47. Метаболизм – совокупность взаимосвязанных биохимических процессов в организме. Наследственные болезни обмена – нарушение метаболизма углеводов, аминокислот,
- 48. Наследственные нарушения метаболизма Мутации в гене, кодирующем фермент приводят к нарушению его работы Метаболизм дерматансульфата MPS
- 49. Когда проявляются наследственные обменные заболевания ? Могут проявляться в любом возрасте от младенческого до взрослого, в
- 50. Наследственные нарушения метаболизма – редкие заболевания
- 51. Наследственные болезни обмена веществ часто сочетаются с инфекционными заболеваниями ! Диагностика возможна только с помощью лабораторных
- 52. Классификация врожденных ошибок метаболизма НБО: протекающие с интоксикацией при которых страдает энергообмен нарушения синтеза или катаболизма
- 53. Заболевания, протекающие с интоксикацией: 1. Острое течение. Проявляются развитием метаболических кризов и могут развиваться как «неонатальные
- 54. НБО, протекающие с интоксикацией: Аминоацидопатии (ФКУ и др.) Органические ацидурии (Большая их часть) Нарушения цикла мочевины
- 55. Заболевания вследствие нарушения синтеза или катаболизма сложных молекул Прогрессирующее течение Клиническое течение не зависит от состава
- 56. Нормальный катаболизм Субстрат Нарушение активности фермента Накопление субстрата блокированной реакции Патогенез ЛБН
- 57. Лизосомные болезни накопления
- 58. Лизосомные болезни накопления
- 59. Лизосомные болезни накопления
- 60. Лизосомные болезни накопления
- 61. МПС I, характеристика заболевания
- 62. Что представляет собой болезнь Фабри ? Врожденное (Х-сцепленное) обменное заболевание Относится к группе, состоящей из более
- 63. Схема наследования Больные лица мужского пола передают дефектный ген всем дочерям и никому из сыновей Разъединение
- 64. Схема наследования У женщин-носителей вероятность передать патологический ген потомству составляет 50% для каждой беременности Разъединение Х-сцепленного
- 65. Полисистемные проявления Связаны с лежащим в основе заболевания поражением различных типов клеток и параллельно сосудистой и
- 66. Личные наблюдения
- 68. Болевой синдром Акропарестезия носит постоянный характер локализация - кисти и стопы ощущается, как жжение, покалывание, боль
- 69. Диагностика ЛБН Генотип Белок Метаболиты Клинический фенотип Биохимический фенотип ДНК-диагностика Энзимодиагностика Анализ метаболитов Клиническая диагностика
- 70. Варианты аутосомно-доминантной гиперхолестеринемии Семейная гиперхолестеринемия (FH, связанная с мутациями рецептора для ЛПНП) 1:500 FDB (свяанная с
- 71. Родословная ребенка М-а, 16 лет с гиперхолестеринемией
- 72. Ксантомы у ребенка с ГЛП IIa типа
- 73. Ксантомы у ребенка с ГЛП IIa типа
- 74. МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
- 75. ГЕНЫ МИТОХОНДРИАЛЬНОЙ ДНК 13 ГЕНОВ КОДИРУЮТ БЕЛКИ ЧЕТЫРЕХ КОМПЛЕКСОВ ДЫХАТЕЛЬНЫХ ЦЕПЕЙ МИТОХОНДРИЙ (1,3,4,5) 22 ГЕНА ТРАНСПОРТНОЙ
- 76. ГРУППЫ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ Заболевания, обусловленные мутациями в митохондриальном геноме Заболевания, обусловленные мутациями в генах ядерной ДНК
- 77. Общие закономерности митохондриальных заболеваний с мутациями в мтДНК Материнский тип наследования Феномен гетероплазмии: случайное распределение копий
- 78. Общие закономерности митохондриальных заболеваний с мутациями в мтДНК прогрессирование с возрастом, что коррелирует с возрастным снижением
- 79. Интенсивно протекающие в митохондриях окислительно-восстановительные процессы с избытком поставляют свободные радикалы, повреждающие ДНК. Митохондриальная ДНК, в
- 80. При слиянии мужской и женской половых клеток зигота получает практически все свои митохондрии (примерно 2х103), а
- 81. Феномен гетероплазмии – результате присутствия в пределах одной клетки митохондрий с разными вариантами геномов. Человек с
- 82. пороговый эффект: для возникновения серьезных нарушений энергетического обмена и дисфункции конкретного органа или ткани необходимо минимальное
- 83. Симптомы митохондриальных заболеваний Повторные коматозные состояния, сопровождающиеся кетоацидозом и гиперкетонурией Задержка физического развития Миопатии и кардиомиопатии
- 84. Симптомы митохондриальных заболеваний Дисфункция щитовидной железы Тубулопатии, витами Д резистентный рахит Диарея, целиакия-подобный синдром Атрофия зрительных
- 85. Клинические фенотипы митохондриальных заболеваний Синдром Кернс-Сейра Прогрессирующая наружная офтальмоплегия (РЕО) Злокачественная мигрень Синдром MELAS Cиндром NARP
- 86. Атрофия дисков зрительных нервов Лебера Манифестация 12-30 лет. Чаще поражаются мужчины. Острая безболезненная потеря зрения на
- 87. Синдром MELAS материнский тип наследования, манифестация в 5-15 лет, возраст проявления болезни - до 40 лет,
- 88. Синдром MELAS инсультоподобные эпизоды с наличием изменений на MRT головного мозга. лактат-ацидоз рваные красные волокна в
- 89. МРТ головного мозга при синдроме MELAS
- 90. Частота встречаемости мутаций в мтДНК при синдроме MELAS в гене MTTL1 3243A>G – 80% 3271T>C -7,5%
- 91. Заболевания множественных делеций и деплеций митохондриальной ДНК.
- 92. Фенотипы мутации в гене РОLG1 Синдром Альперса ( начало на 1 году жизни, спастика, судороги, задержка
- 93. Окулофарингеальная миопатия
- 94. Биохимическая диагностика Увеличение концентрации лактата в крови при физической нагрузке или после нагрузки глюкозой Парадоксальная гиперкетонемия,
- 95. Морфологическая диагностика митохондриальных заболеваний В биоптате мышечных волокон- феномен RRF ( rigger red fibers) при окраске
- 96. Молекулярно-генетическая диагностика Методы NGS Цифровая ПЦР
- 98. Эволюция представлений о моногенных болезнях Один ген – одна болезнь Различные мутации в одном гене определяют
- 99. Эволюция представлений о моногенных болезнях Один фенотип – много генов Один фенотип – определенные мутации в
- 101. Скачать презентацию

Генетика - наука о наследственности
и изменчивости.
Термин ввел в 1907 году
Генетика - наука о наследственности
и изменчивости.
Термин ввел в 1907 году

Генетика человека - фундаментальная наука
Часть общей генетики, изучающей законы накопления, передачи
Генетика человека - фундаментальная наука
Часть общей генетики, изучающей законы накопления, передачи

Медицинская генетика - наука о роли наследственности в патологии человека, закономерностях
Медицинская генетика - наука о роли наследственности в патологии человека, закономерностях

Развитие генетики человека
1952 T.C. Hsu Открытие гипотонической обработки
Дж. Уотсон, Ф.
Развитие генетики человека
1952 T.C. Hsu Открытие гипотонической обработки
Дж. Уотсон, Ф.

Развитие генетики человека
Первый автоматический секвенатор, 1987 г.
1993 год Кэрри Мюллис Нобелевская
Развитие генетики человека
Первый автоматический секвенатор, 1987 г.
1993 год Кэрри Мюллис Нобелевская

Развитие клинической генетики
1959 – Франсуа Лежен
47 хромосом (трисомия 21)
Развитие клинической генетики
1959 – Франсуа Лежен
47 хромосом (трисомия 21)

Успехи клинической генетики в СССР
С.И. Давиденков
В 1925 г.
Успехи клинической генетики в СССР
С.И. Давиденков
В 1925 г.

В Москве в 1935 г был создан «Медико-генетический институт». Соломон Григорьевич

Развитие клинической генетики в СССР
Постановлением Президиума АМН СССР 01.03.61 была создана
Развитие клинической генетики в СССР
Постановлением Президиума АМН СССР 01.03.61 была создана

Основные нормативные документы, регулирующие вопросы редких заболеваний в РФ
Федеральный закон
Основные нормативные документы, регулирующие вопросы редких заболеваний в РФ
Федеральный закон

Всемирная организация здравоохранения (ВОЗ) определяет РЗ следующим образом:
1. Редко встречаются в
Всемирная организация здравоохранения (ВОЗ) определяет РЗ следующим образом:
1. Редко встречаются в

Основные нормативные документы, регулирующие вопросы редких заболеваний в РФ
Постановление Правительства РФ
Основные нормативные документы, регулирующие вопросы редких заболеваний в РФ
Постановление Правительства РФ

ПЕРЕЧЕНЬ ЖИЗНЕУГРОЖАЮЩИХ И ХРОНИЧЕСКИХ ПРОГРЕССИРУЮЩИХ РЕДКИХ (ОРФАННЫХ) ЗАБОЛЕВАНИЙ, ПРИВОДЯЩИХ К СОКРАЩЕНИЮ
ПЕРЕЧЕНЬ ЖИЗНЕУГРОЖАЮЩИХ И ХРОНИЧЕСКИХ ПРОГРЕССИРУЮЩИХ РЕДКИХ (ОРФАННЫХ) ЗАБОЛЕВАНИЙ, ПРИВОДЯЩИХ К СОКРАЩЕНИЮ

ПЕРЕЧЕНЬ ЖИЗНЕУГРОЖАЮЩИХ И ХРОНИЧЕСКИХ ПРОГРЕССИРУЮЩИХ РЕДКИХ (ОРФАННЫХ) ЗАБОЛЕВАНИЙ, ПРИВОДЯЩИХ К СОКРАЩЕНИЮ
ПЕРЕЧЕНЬ ЖИЗНЕУГРОЖАЮЩИХ И ХРОНИЧЕСКИХ ПРОГРЕССИРУЮЩИХ РЕДКИХ (ОРФАННЫХ) ЗАБОЛЕВАНИЙ, ПРИВОДЯЩИХ К СОКРАЩЕНИЮ

Изменения в перечне 24-х нозологий
В 2019 году программа «7 высоко-затратных
Изменения в перечне 24-х нозологий
В 2019 году программа «7 высоко-затратных
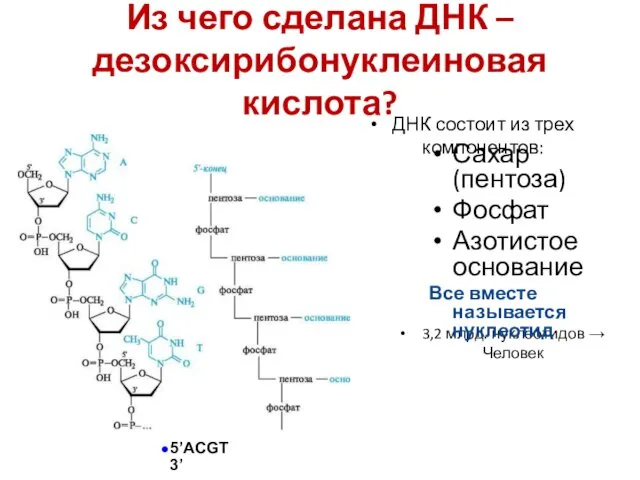
Из чего сделана ДНК – дезоксирибонуклеиновая кислота?
3,2 млрд. нуклеотидов → Человек
Сахар
Из чего сделана ДНК – дезоксирибонуклеиновая кислота?
3,2 млрд. нуклеотидов → Человек
Сахар
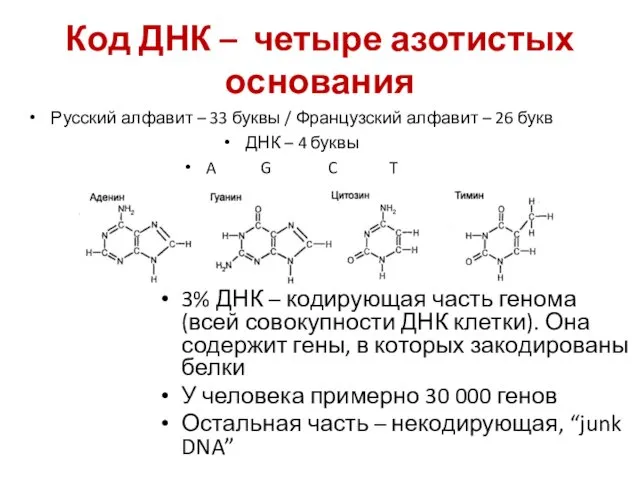
Код ДНК – четыре азотистых основания
3% ДНК – кодирующая часть генома
Код ДНК – четыре азотистых основания
3% ДНК – кодирующая часть генома
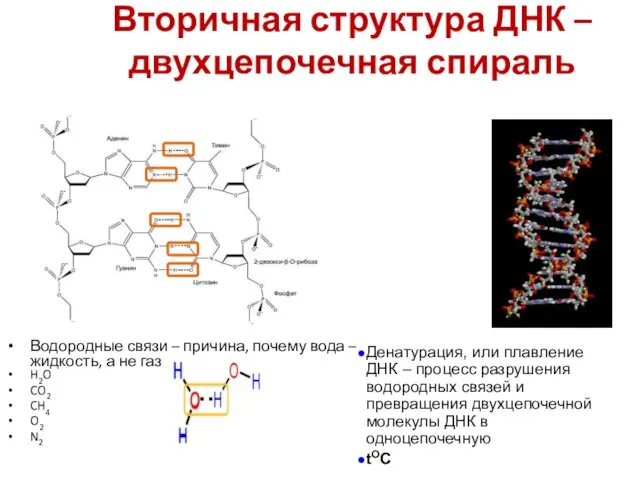
Вторичная структура ДНК – двухцепочечная спираль
A = T
G ≡ C
H2O
CO2
CH4
O2
N2
Водородные
Вторичная структура ДНК – двухцепочечная спираль
A = T
G ≡ C
H2O
CO2
CH4
O2
N2
Водородные
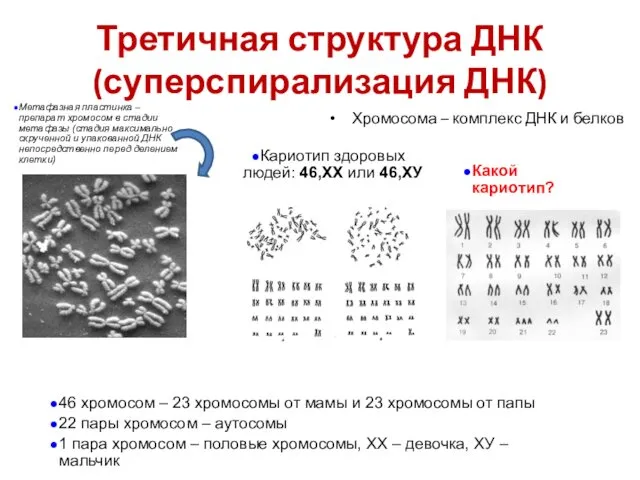
Третичная структура ДНК (суперспирализация ДНК)
Хромосома – комплекс ДНК и белков
46 хромосом
Третичная структура ДНК (суперспирализация ДНК)
Хромосома – комплекс ДНК и белков
46 хромосом
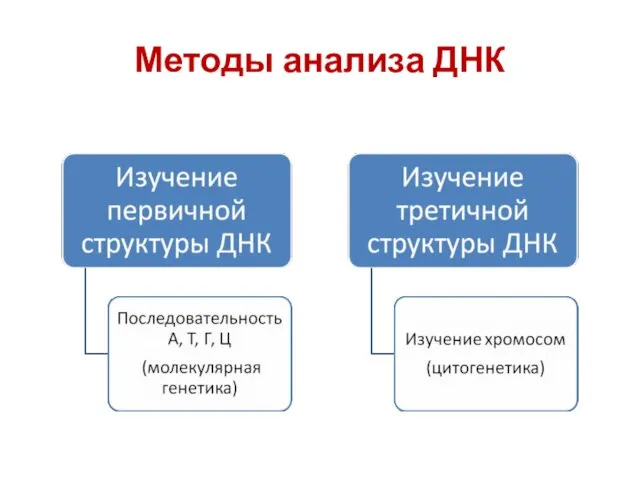
Методы анализа ДНК
Методы анализа ДНК

Структурные элементы гена
Экзон
размер экзома человека — ~ 40 млн.н.п.
Интрон
Экзон-интронная
Структурные элементы гена
Экзон
размер экзома человека — ~ 40 млн.н.п.
Интрон
Экзон-интронная

Распространенные виды мутаций
Миссенс мутация
Нонсенс мутация
Молчащая мутация
Мутация сдвига рамки считывания
Мутация сайта сплайсинга
Распространенные виды мутаций
Миссенс мутация
Нонсенс мутация
Молчащая мутация
Мутация сдвига рамки считывания
Мутация сайта сплайсинга

Методы анализа определения геномных вариантов
ПДАФ (полиморфизм длин амплифицированных фрагментов)
ПДРФ (полиморфизм
Методы анализа определения геномных вариантов
ПДАФ (полиморфизм длин амплифицированных фрагментов)
ПДРФ (полиморфизм

Виды геномных вариантов
Мутация – патогенный геномный вариант
Variant of unknown significance (вариант
Виды геномных вариантов
Мутация – патогенный геномный вариант
Variant of unknown significance (вариант
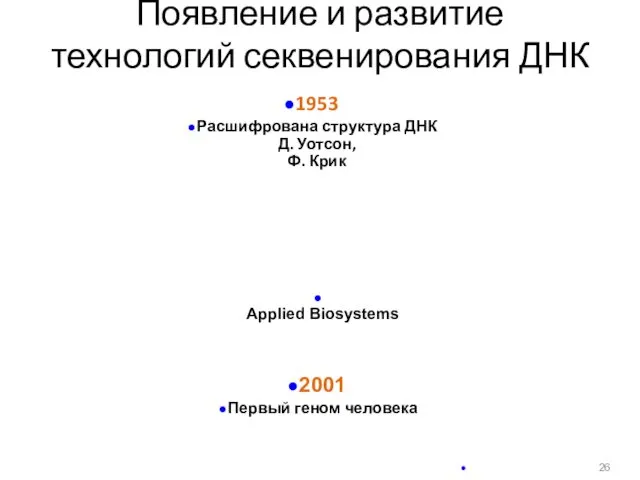
Появление и развитие
технологий секвенирования ДНК
1953
Расшифрована структура ДНК
Д.
Появление и развитие
технологий секвенирования ДНК
1953
Расшифрована структура ДНК
Д.

Классификация наследственных болезней
Хромосомные болезни, связанные с изменением их числа
Хромосомные болезни, связанные
Классификация наследственных болезней
Хромосомные болезни, связанные с изменением их числа
Хромосомные болезни, связанные

Классификация наследственных болезней
Генные болезни, связанные с точечными мутациями ядерной ДНК:
Моногенные заболевания,
Классификация наследственных болезней
Генные болезни, связанные с точечными мутациями ядерной ДНК:
Моногенные заболевания,

Классификация наследственных болезней
Болезни, обусловленные дефектами митохондриальной ДНК:
Генные, связанные с точечными мутациями
Классификация наследственных болезней
Болезни, обусловленные дефектами митохондриальной ДНК:
Генные, связанные с точечными мутациями

Хромосомные болезни
Числовые аномалии хромосом
~
Хромосомные болезни
Числовые аномалии хромосом
~

Хромосомные болезни
Структурные аномалии хромосом
~ 1 из 500 индивидов является носителем структурной
Хромосомные болезни
Структурные аномалии хромосом
~ 1 из 500 индивидов является носителем структурной

Структурные аномалии хромосом
транслокации
делеции
дупликации
инверсии
инсерции
кольцевые хромосомы
маркерные хромосомы
Структурные аномалии хромосом
транслокации
делеции
дупликации
инверсии
инсерции
кольцевые хромосомы
маркерные хромосомы
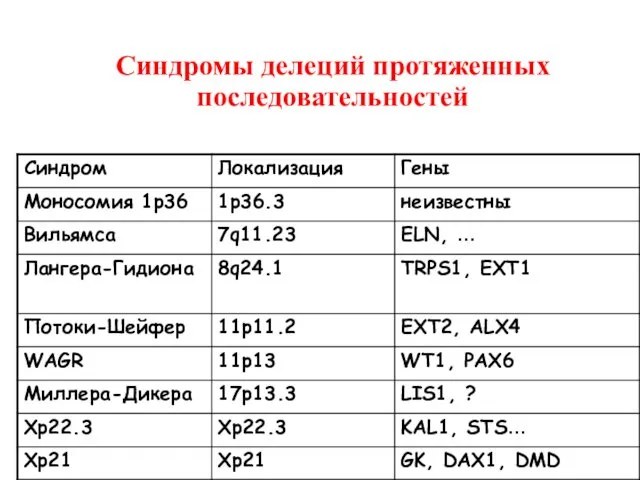
Синдромы делеций протяженных последовательностей
Синдромы делеций протяженных последовательностей

Делеция 11p11.2p12
(синдром Потоки-Шейфер)
OMIM# 601224
Отставание в умственном развитии
Множественные экзостозы (EXT2 )
Теменные окна
Делеция 11p11.2p12
(синдром Потоки-Шейфер)
OMIM# 601224
Отставание в умственном развитии
Множественные экзостозы (EXT2 )
Теменные окна
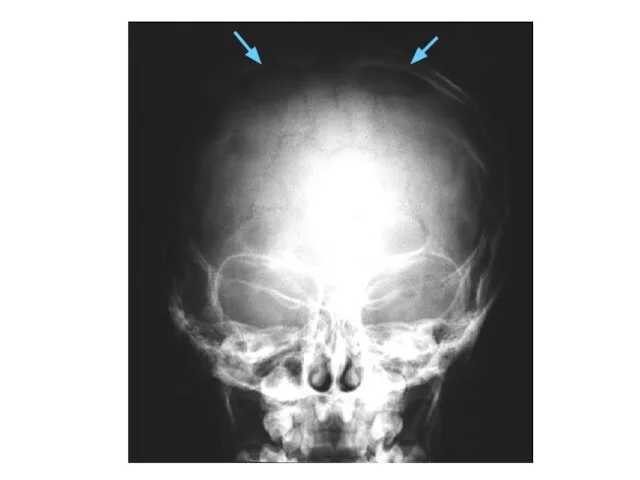
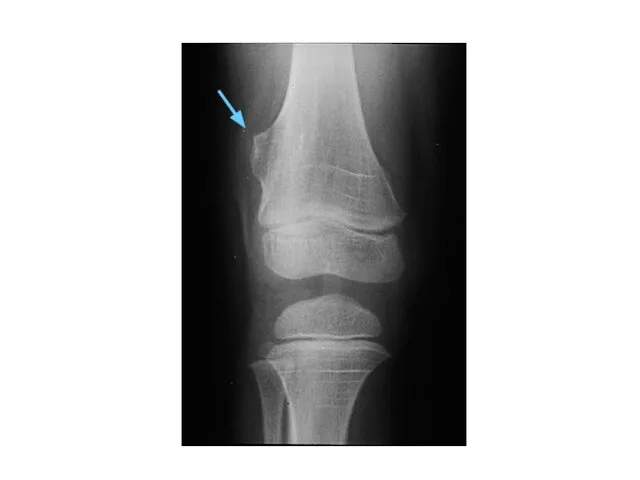

Синдром Вильямса
deletion 7q11.23q11.23
OMIM# 194050
Отставание в умственном развитии
ВПС (75%) SVAS
Гиперкальцемия
Лицевые микроаномалии
Синдром Вильямса
deletion 7q11.23q11.23
OMIM# 194050
Отставание в умственном развитии
ВПС (75%) SVAS
Гиперкальцемия
Лицевые микроаномалии


Методы диагностики хромосомных заболеваний
Клинические - выявление микроаномалий развития и пороков
Методы диагностики хромосомных заболеваний
Клинические - выявление микроаномалий развития и пороков
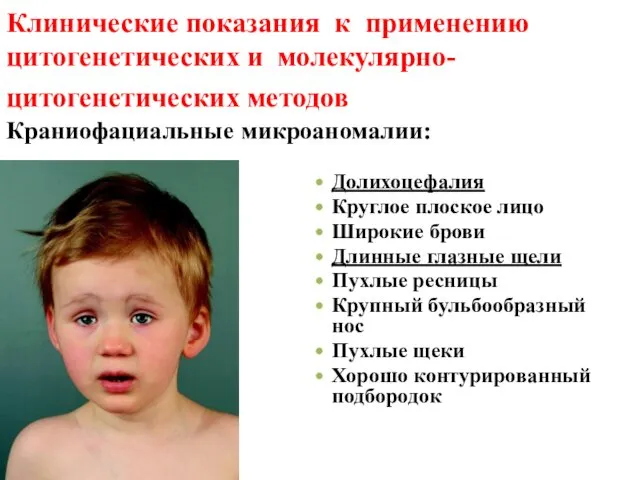
Клинические показания к применению цитогенетических и молекулярно- цитогенетических методов Краниофациальные
Клинические показания к применению цитогенетических и молекулярно- цитогенетических методов Краниофациальные

Клинические показания к применению цитогенетических и молекулярно- цитогенетических методов
Неспецифическая умственная
Клинические показания к применению цитогенетических и молекулярно- цитогенетических методов
Неспецифическая умственная
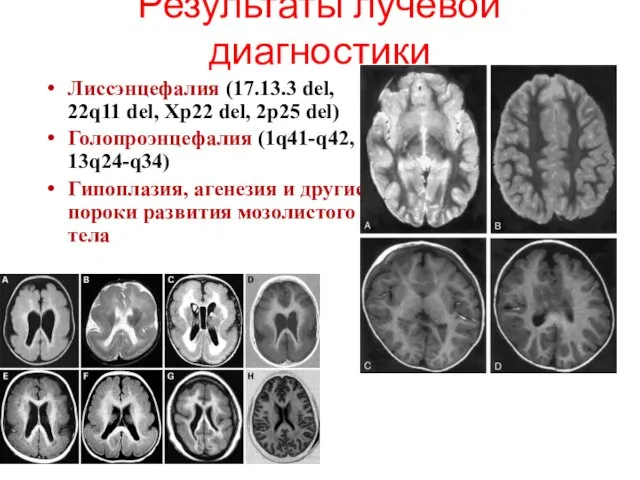
Результаты лучевой диагностики
Лиссэнцефалия (17.13.3 del, 22q11 del, Xp22 del, 2p25
Результаты лучевой диагностики
Лиссэнцефалия (17.13.3 del, 22q11 del, Xp22 del, 2p25

Генные болезни
Связанные с точечными мутациями ядерной ДНК:
моногенные заболевания, связанные
Генные болезни
Связанные с точечными мутациями ядерной ДНК:
моногенные заболевания, связанные

Типы наследования моногенных заболеваний
Аутосомно-рецессивный (большинство заболеваний)
Аутосомно-доминантный
Х-сцепленный рецессивный
Митохондриальный
Типы наследования моногенных заболеваний
Аутосомно-рецессивный (большинство заболеваний)
Аутосомно-доминантный
Х-сцепленный рецессивный
Митохондриальный

Сложности клинического этапа диагностики моногенных заболеваний
Наследственные заболевания редкие, представленные единичными
Сложности клинического этапа диагностики моногенных заболеваний
Наследственные заболевания редкие, представленные единичными
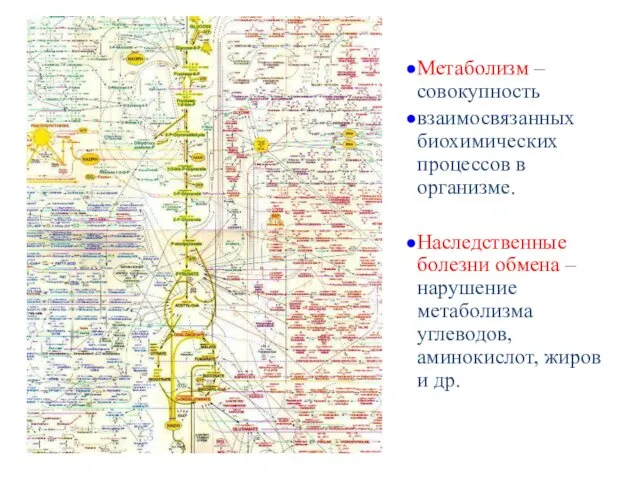
Метаболизм – совокупность
взаимосвязанных биохимических процессов в организме.
Наследственные болезни обмена – нарушение
Метаболизм – совокупность
взаимосвязанных биохимических процессов в организме.
Наследственные болезни обмена – нарушение
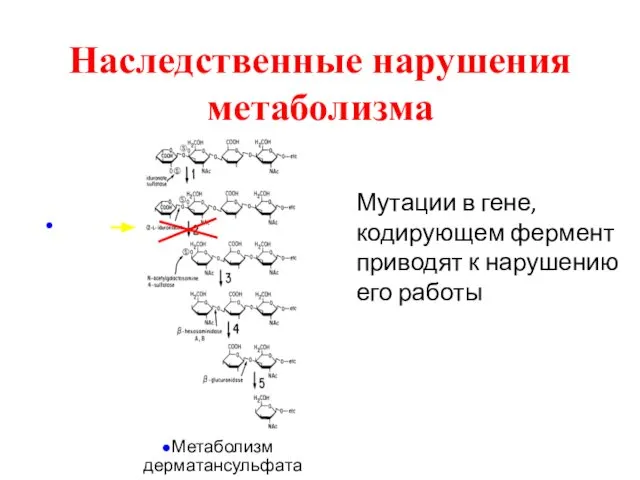
Наследственные нарушения метаболизма
Мутации в гене, кодирующем фермент приводят к нарушению его
Наследственные нарушения метаболизма
Мутации в гене, кодирующем фермент приводят к нарушению его

Когда проявляются наследственные обменные заболевания ?
Могут проявляться в любом возрасте от
Когда проявляются наследственные обменные заболевания ?
Могут проявляться в любом возрасте от

Наследственные нарушения метаболизма – редкие заболевания
Наследственные нарушения метаболизма – редкие заболевания

Наследственные болезни обмена веществ часто сочетаются с инфекционными заболеваниями !
Диагностика возможна
Наследственные болезни обмена веществ часто сочетаются с инфекционными заболеваниями !
Диагностика возможна

Классификация врожденных ошибок метаболизма
НБО:
протекающие с интоксикацией
при которых
Классификация врожденных ошибок метаболизма
НБО:
протекающие с интоксикацией
при которых

Заболевания, протекающие с интоксикацией:
1. Острое течение. Проявляются развитием метаболических
Заболевания, протекающие с интоксикацией:
1. Острое течение. Проявляются развитием метаболических

НБО, протекающие с интоксикацией:
Аминоацидопатии (ФКУ и др.)
Органические ацидурии (Большая их часть)
Нарушения
НБО, протекающие с интоксикацией:
Аминоацидопатии (ФКУ и др.)
Органические ацидурии (Большая их часть)
Нарушения

Заболевания вследствие нарушения синтеза или катаболизма сложных молекул
Прогрессирующее течение
Клиническое
Заболевания вследствие нарушения синтеза или катаболизма сложных молекул
Прогрессирующее течение
Клиническое
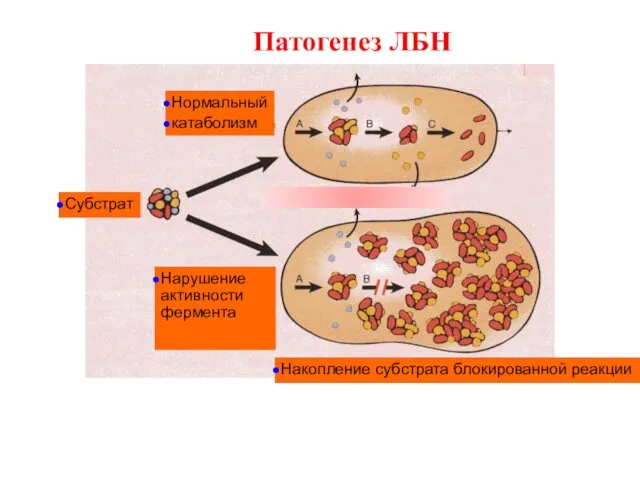
Нормальный
катаболизм
Субстрат
Нарушение активности фермента
Накопление субстрата блокированной реакции
Патогенез ЛБН
Нормальный
катаболизм
Субстрат
Нарушение активности фермента
Накопление субстрата блокированной реакции
Патогенез ЛБН
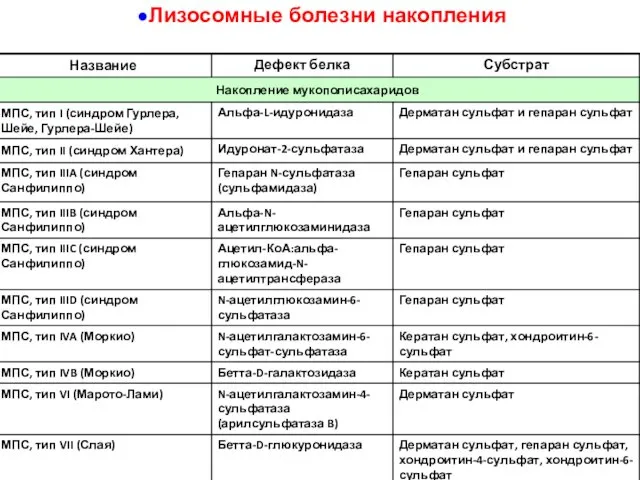
Лизосомные болезни накопления
Лизосомные болезни накопления
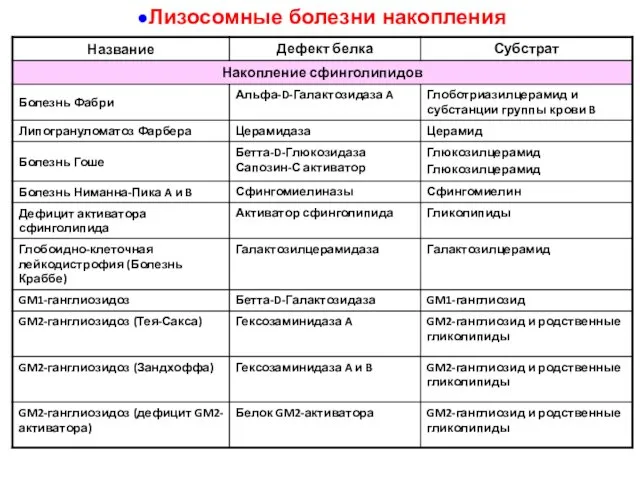
Лизосомные болезни накопления
Лизосомные болезни накопления
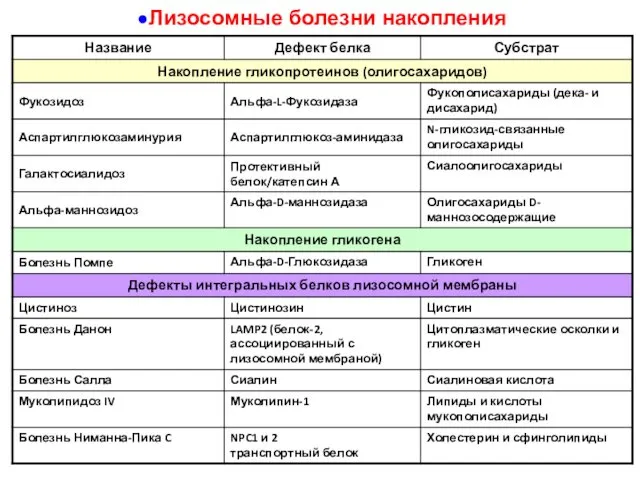
Лизосомные болезни накопления
Лизосомные болезни накопления
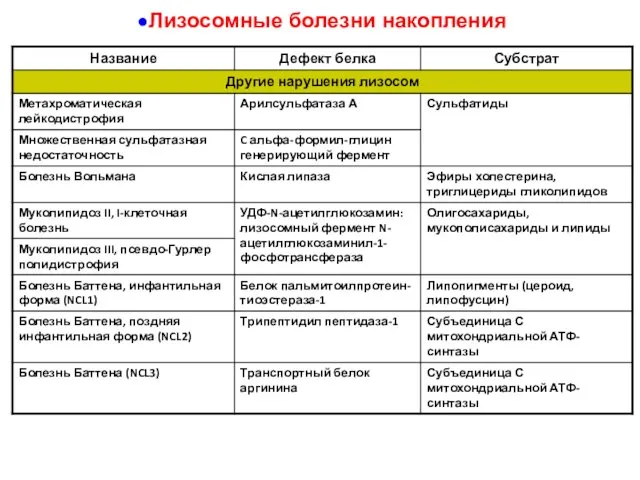
Лизосомные болезни накопления
Лизосомные болезни накопления
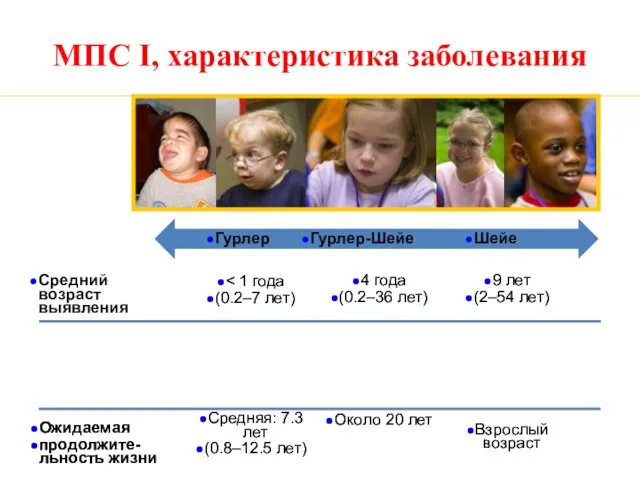
МПС I, характеристика заболевания
МПС I, характеристика заболевания

Что представляет собой болезнь Фабри ?
Врожденное (Х-сцепленное) обменное заболевание
Относится к группе,
Что представляет собой болезнь Фабри ?
Врожденное (Х-сцепленное) обменное заболевание
Относится к группе,
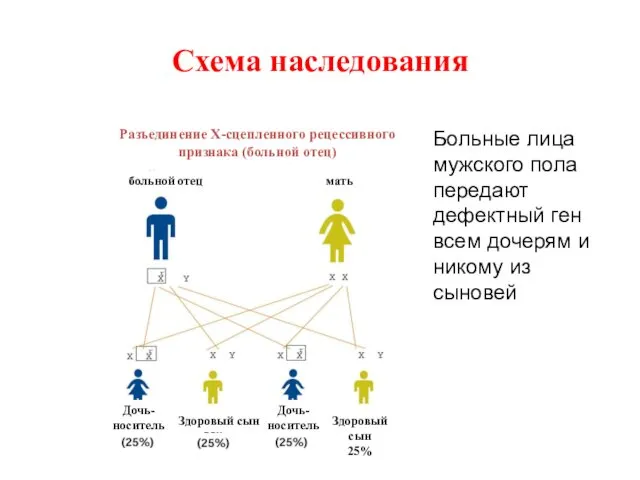
Схема наследования
Больные лица мужского пола передают дефектный ген всем дочерям и
Схема наследования
Больные лица мужского пола передают дефектный ген всем дочерям и

Схема наследования
У женщин-носителей вероятность передать патологический ген потомству составляет 50% для
Схема наследования
У женщин-носителей вероятность передать патологический ген потомству составляет 50% для

Полисистемные проявления
Связаны с лежащим в основе заболевания поражением различных типов клеток
Полисистемные проявления
Связаны с лежащим в основе заболевания поражением различных типов клеток
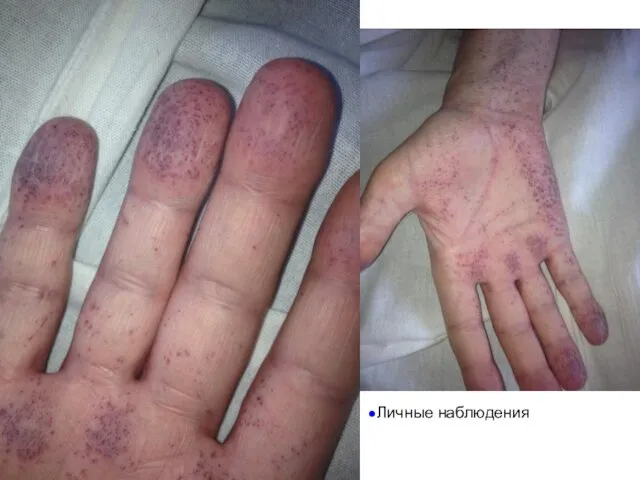
Личные наблюдения
Личные наблюдения
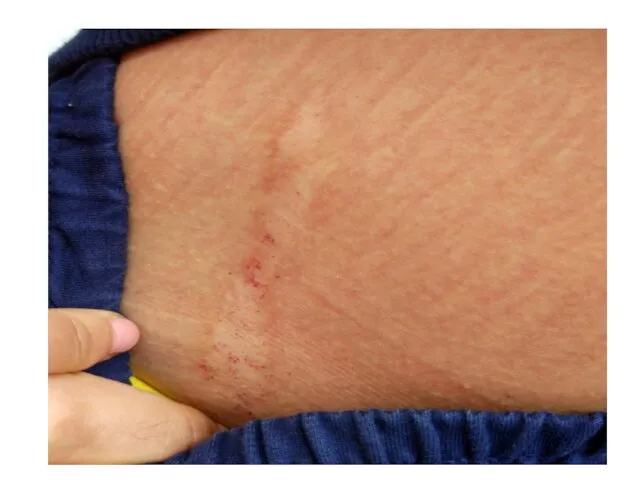

Болевой синдром
Акропарестезия
носит постоянный характер
локализация - кисти и стопы
ощущается, как жжение, покалывание,
Болевой синдром
Акропарестезия
носит постоянный характер
локализация - кисти и стопы
ощущается, как жжение, покалывание,
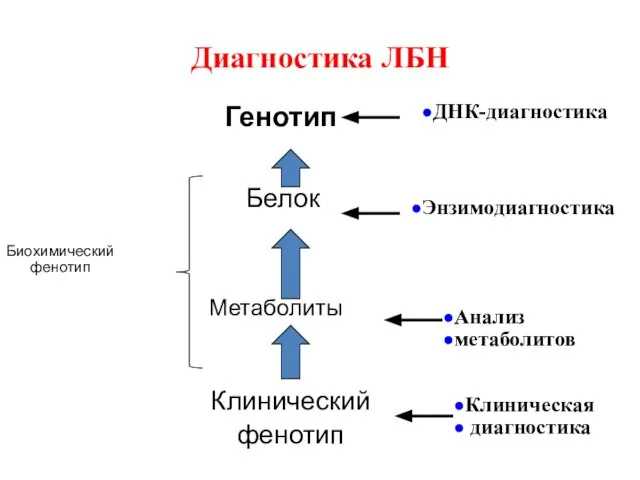
Диагностика ЛБН
Генотип
Белок
Метаболиты
Клинический
фенотип
Биохимический фенотип
ДНК-диагностика
Энзимодиагностика
Анализ
метаболитов
Клиническая
диагностика
Диагностика ЛБН
Генотип
Белок
Метаболиты
Клинический
фенотип
Биохимический фенотип
ДНК-диагностика
Энзимодиагностика
Анализ
метаболитов
Клиническая
диагностика

Варианты аутосомно-доминантной гиперхолестеринемии
Семейная гиперхолестеринемия (FH, связанная с мутациями рецептора для ЛПНП)
Варианты аутосомно-доминантной гиперхолестеринемии
Семейная гиперхолестеринемия (FH, связанная с мутациями рецептора для ЛПНП)
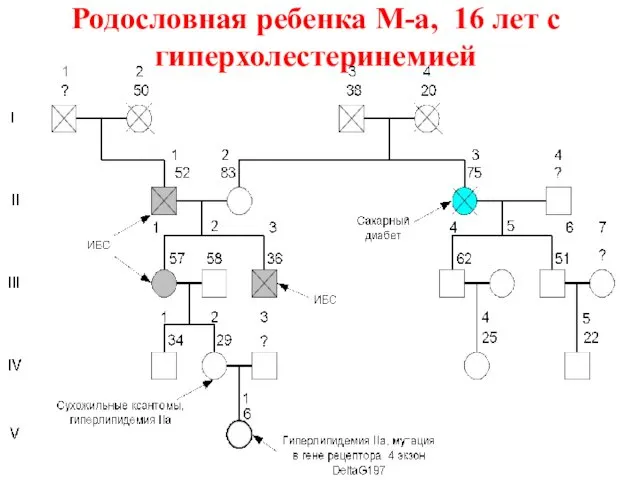
Родословная ребенка М-а, 16 лет с гиперхолестеринемией
Родословная ребенка М-а, 16 лет с гиперхолестеринемией

Ксантомы у ребенка с ГЛП IIa типа
Ксантомы у ребенка с ГЛП IIa типа
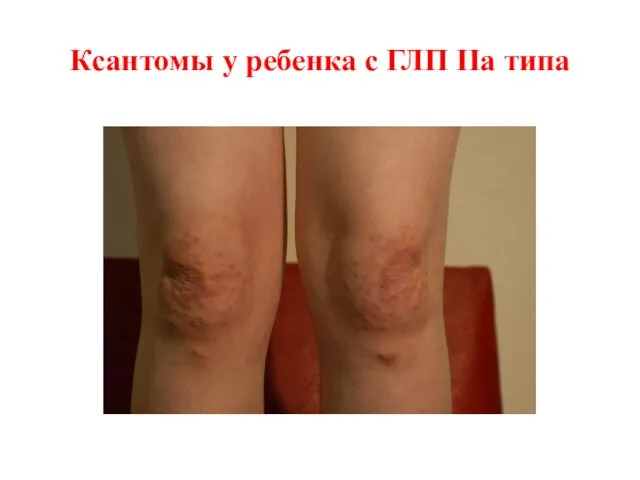
Ксантомы у ребенка с ГЛП IIa типа
Ксантомы у ребенка с ГЛП IIa типа

МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ

ГЕНЫ МИТОХОНДРИАЛЬНОЙ ДНК
13 ГЕНОВ КОДИРУЮТ БЕЛКИ ЧЕТЫРЕХ КОМПЛЕКСОВ ДЫХАТЕЛЬНЫХ ЦЕПЕЙ МИТОХОНДРИЙ
ГЕНЫ МИТОХОНДРИАЛЬНОЙ ДНК
13 ГЕНОВ КОДИРУЮТ БЕЛКИ ЧЕТЫРЕХ КОМПЛЕКСОВ ДЫХАТЕЛЬНЫХ ЦЕПЕЙ МИТОХОНДРИЙ

ГРУППЫ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Заболевания, обусловленные мутациями в митохондриальном геноме
Заболевания, обусловленные мутациями в
ГРУППЫ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Заболевания, обусловленные мутациями в митохондриальном геноме
Заболевания, обусловленные мутациями в

Общие закономерности митохондриальных заболеваний с мутациями в мтДНК
Материнский тип наследования
Феномен гетероплазмии:
Общие закономерности митохондриальных заболеваний с мутациями в мтДНК
Материнский тип наследования
Феномен гетероплазмии:

Общие закономерности митохондриальных заболеваний с мутациями в мтДНК
прогрессирование с возрастом, что
Общие закономерности митохондриальных заболеваний с мутациями в мтДНК
прогрессирование с возрастом, что

Интенсивно протекающие в митохондриях окислительно-восстановительные процессы с избытком поставляют свободные
Интенсивно протекающие в митохондриях окислительно-восстановительные процессы с избытком поставляют свободные

При слиянии мужской и женской половых клеток зигота получает практически все
При слиянии мужской и женской половых клеток зигота получает практически все

Феномен гетероплазмии – результате присутствия в пределах одной клетки митохондрий
Феномен гетероплазмии – результате присутствия в пределах одной клетки митохондрий

пороговый эффект: для возникновения серьезных нарушений энергетического обмена и дисфункции конкретного
пороговый эффект: для возникновения серьезных нарушений энергетического обмена и дисфункции конкретного

Симптомы митохондриальных заболеваний
Повторные коматозные состояния, сопровождающиеся кетоацидозом и гиперкетонурией
Задержка физического развития
Миопатии
Симптомы митохондриальных заболеваний
Повторные коматозные состояния, сопровождающиеся кетоацидозом и гиперкетонурией
Задержка физического развития
Миопатии

Симптомы митохондриальных заболеваний
Дисфункция щитовидной железы
Тубулопатии, витами Д резистентный рахит
Диарея, целиакия-подобный синдром
Атрофия
Симптомы митохондриальных заболеваний
Дисфункция щитовидной железы
Тубулопатии, витами Д резистентный рахит
Диарея, целиакия-подобный синдром
Атрофия

Клинические фенотипы митохондриальных заболеваний
Синдром Кернс-Сейра
Прогрессирующая наружная офтальмоплегия (РЕО)
Злокачественная мигрень
Синдром MELAS
Cиндром NARP
Синдром
Клинические фенотипы митохондриальных заболеваний
Синдром Кернс-Сейра
Прогрессирующая наружная офтальмоплегия (РЕО)
Злокачественная мигрень
Синдром MELAS
Cиндром NARP
Синдром

Атрофия дисков зрительных нервов Лебера
Манифестация 12-30 лет. Чаще поражаются мужчины.
Острая безболезненная
Атрофия дисков зрительных нервов Лебера
Манифестация 12-30 лет. Чаще поражаются мужчины.
Острая безболезненная

Синдром MELAS
материнский тип наследования,
манифестация в 5-15 лет, возраст проявления болезни -
Синдром MELAS
материнский тип наследования,
манифестация в 5-15 лет, возраст проявления болезни -

Синдром MELAS
инсультоподобные эпизоды с наличием изменений на MRT головного мозга.
лактат-ацидоз
рваные
Синдром MELAS
инсультоподобные эпизоды с наличием изменений на MRT головного мозга.
лактат-ацидоз
рваные
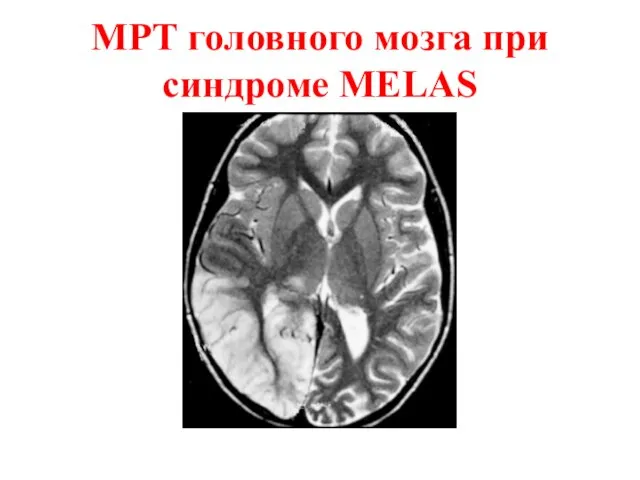
МРТ головного мозга при синдроме MELAS
МРТ головного мозга при синдроме MELAS

Частота встречаемости мутаций в мтДНК при синдроме MELAS в
гене MTTL1
3243A>G –
Частота встречаемости мутаций в мтДНК при синдроме MELAS в
гене MTTL1
3243A>G –
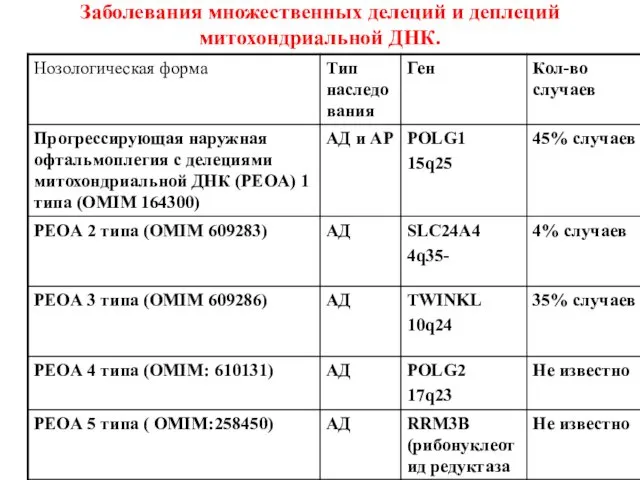
Заболевания множественных делеций и деплеций митохондриальной ДНК.
Заболевания множественных делеций и деплеций митохондриальной ДНК.

Фенотипы мутации в гене РОLG1
Синдром Альперса ( начало на 1 году
Фенотипы мутации в гене РОLG1
Синдром Альперса ( начало на 1 году
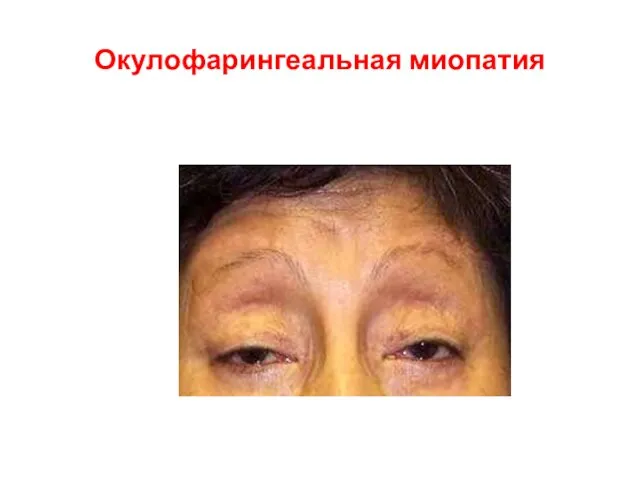
Окулофарингеальная миопатия
Окулофарингеальная миопатия

Биохимическая диагностика
Увеличение концентрации лактата в крови при физической нагрузке или после
Биохимическая диагностика
Увеличение концентрации лактата в крови при физической нагрузке или после

Морфологическая диагностика митохондриальных заболеваний
В биоптате мышечных волокон- феномен RRF ( rigger
Морфологическая диагностика митохондриальных заболеваний
В биоптате мышечных волокон- феномен RRF ( rigger

Молекулярно-генетическая диагностика
Методы NGS
Цифровая ПЦР
Молекулярно-генетическая диагностика
Методы NGS
Цифровая ПЦР

Эволюция представлений о моногенных болезнях
Один ген – одна болезнь
Различные мутации в
Эволюция представлений о моногенных болезнях
Один ген – одна болезнь
Различные мутации в

Эволюция представлений о моногенных болезнях
Один фенотип – много генов
Один фенотип –
Эволюция представлений о моногенных болезнях
Один фенотип – много генов
Один фенотип –
 Диарейный синдром в клинике инфекционных болезней. Патогенез и клинические проявления. Принципы лечения дегидратационного шока
Диарейный синдром в клинике инфекционных болезней. Патогенез и клинические проявления. Принципы лечения дегидратационного шока Пластические операции в маммологии
Пластические операции в маммологии Грыжа пищеводного отверстия диафрагмы
Грыжа пищеводного отверстия диафрагмы Viral hepatitis
Viral hepatitis Методы диагностики в неврологии
Методы диагностики в неврологии Аппараты ингаляционного наркоза: новые тенденции
Аппараты ингаляционного наркоза: новые тенденции Вирустық инфекцияның химиотерапиялық негіздері
Вирустық инфекцияның химиотерапиялық негіздері Bronchitis
Bronchitis Синдром Бругада
Синдром Бругада Дополнительные методы обследования в клинике ортопедической стоматологии. Диагноз, дифференциальный диагноз
Дополнительные методы обследования в клинике ортопедической стоматологии. Диагноз, дифференциальный диагноз Интенсивная терапия тяжелой преэклампсии и эклампсии (позиция доказательной медицины)
Интенсивная терапия тяжелой преэклампсии и эклампсии (позиция доказательной медицины) Мұрынның тампонадасы
Мұрынның тампонадасы Антропометрія, як спосіб вимірювання частин тіла спортсмена. Соматотипування
Антропометрія, як спосіб вимірювання частин тіла спортсмена. Соматотипування Глюкокортикордты остеопороз
Глюкокортикордты остеопороз Общие и местные факторы риска возникновения кариеса
Общие и местные факторы риска возникновения кариеса Опухоли из эпителия. Рак отдельных локализаций (молочной железы, матки)
Опухоли из эпителия. Рак отдельных локализаций (молочной железы, матки) Общие принципы диагностики в травматологии и ортопедии
Общие принципы диагностики в травматологии и ортопедии Трансплантация тонкой кишки
Трансплантация тонкой кишки Адаптация первоклассников. Пути преодоления дезадаптации
Адаптация первоклассников. Пути преодоления дезадаптации ГБУЗ Челябинская областная детская клиническая больница
ГБУЗ Челябинская областная детская клиническая больница Bacterial infections. Disorders of hair
Bacterial infections. Disorders of hair СПИД . Прошлое, настоящее, будущее!
СПИД . Прошлое, настоящее, будущее! Кашель при ОРЗ: рациональная тактика лечения на педиатрическом участке
Кашель при ОРЗ: рациональная тактика лечения на педиатрическом участке Вирус бешенства
Вирус бешенства Геморрагический шок в гинекологии
Геморрагический шок в гинекологии Особенности общения дошкольников со сверстниками
Особенности общения дошкольников со сверстниками Стоматологическое обследование пациента. 3 курс. Тема №2
Стоматологическое обследование пациента. 3 курс. Тема №2 Сложные и нерешённые вопросы МСЭ в кардиологии
Сложные и нерешённые вопросы МСЭ в кардиологии