- Pharmacology. Lecture 1

Содержание
- 2. Medical pharmacology is the science of chemicals (drugs) that interact with the human body. These interactions
- 3. Routes of administration: ADVANTAGES and DISADVANTAGES Oral: the most common and safest, convenient, and economical route
- 5. Rectal: partially bypasses first-pass effect, destruction by stomach acid, ideal if drug causes vomiting, in patients
- 6. Intravenous: can have immediate effects, ideal if dosed in large volumes, suitable for irritating substances and
- 7. Subcutaneous: suitable for slow-release drugs, ideal for some poorly soluble suspensions, but pain or necrosis if
- 9. Transdermal (patch): bypasses the first-pass effect, convenient and painless, ideal for drugs that are lipophilic and
- 11. Inhalation: absorption is rapid; can have immediate effects, ideal for gases, effective for patients with respiratory
- 13. Mechanisms of absorption of drugs Drugs may be absorbed from the GI tract by passive diffusion,
- 14. Diffusion: lipid-soluble drugs readily move across most biologic membranes due to their solubility in the membrane
- 15. Active transport: Energy-dependent, involves specific carrier proteins. It is capable of moving drugs against a concentration
- 18. Bioavailability Bioavailability is the rate and extent to which an administered drug reaches the systemic circulation.
- 19. Determination of bioavailability: Bioavailability is determined by comparing plasma levels of a drug after a particular
- 21. Factors that influence bioavailability First-pass hepatic metabolism: When a drug is absorbed from the GI tract,
- 22. Chemical instability. Nature of the drug formulation (salt form, crystal polymorphism, enteric coatings, and the presence
- 23. Drug distribution Drug distribution is the process by which a drug reversibly leaves the bloodstream and
- 24. Lipid-soluble drugs readily penetrate the CNS because they dissolve in the endothelial cell membrane. Ionized or
- 27. Elimination Once a drug enters the body, the process of elimination begins. The three major routes
- 29. Metabolism in the liver leads to production of products with increased polarity, which allows the drug
- 31. The CYP450-dependent enzymes are an important target for pharmacokinetic drug interactions. Inducers (for example, phenobarbital, rifampin,
- 32. Phase II: This phase consists of conjugation reactions with an endogenous substrate, such as glucuronic acid,
- 33. Drug clearance may also occur via the intestines, bile, lungs, and breast milk, among others. Drugs
- 34. Pharmacodynamics. Effects Local effect occurs at the site of drug’s application . Resorptive (systemic) effect develops
- 35. Drugs “’targets Few drugs (e.g. activated charcoal, osmotic diuretics) act by virtue of their physicochemical properties,
- 36. Enzymes. Drugs that act by inhibiting enzymes include: anticholinesterases, which enhance the action of acetylcholine; carbonic
- 37. Drugs can influence on ion channels (selective pores in the membrane). Among drugs affecting ion channels
- 38. However, most drugs produce their effects by acting on specific proteins. These proteins are called receptors,
- 39. A receptor as any biologic molecule to which a drug binds and produces a measurable response.
- 42. 3) Kinase‐linked receptors are surface receptors that possess (usually) intrinsic tyrosine kinase activity. They include receptors
- 45. Chemicals (e.g. acetylcholine) or drugs that activate receptors and produce a response are called agonists .
- 46. Partial agonists. These are agonists that cannot elicit the same maximum response as a ‘full’ agonist.
- 49. The durability of the “Drug-receptor” bond determines whether the drug action is reversible (characteristic for most
- 50. Potency is a measure of the amount of drug necessary to produce an effect of a
- 51. Efficacy is the magnitude of response a drug causes when it interacts with a receptor. Efficacy
- 52. Drugs interactions Pharmacokinetic Pharmacodynamic: Synergism Antagonism
- 53. In case of synergism drug interaction leads to an increase in effect. Synergism may be direct
- 54. Interaction of drugs Synergism Summation (paracetamol + metamizol sodium) Potentiation (paracetamol + diphenhydramine)) 1 1 2
- 55. Antagonism The ability of a drug to decrease the effect of the other one is called
- 56. Irreversible antagonists have an effect that cannot be reversed by increasing the concentration of agonist. The
- 57. Synergoantagonism occurs when some effects of the combined drugs are intensified and others are weakened. For
- 58. Pharmacokinetic interactions Absorption. Drugs that increase (e.g. metoclopramide) or decrease (e.g. atropine) the rate of gastric
- 59. Metabolism. Induction of hepatic enzymes by a second drug (e.g.phenobarbital, rifampicin) can decrease the efficacy of
- 60. Adverse drug reactions Adverse drug reactions can be divided into those that are predictable and dose‐related
- 61. Cumulation – storage of pharmacological substance in the body. It is typical for slow-acting drugs, that
- 62. Tachyphylaxis, desensitization, tolerance and drug resistance When a drug is given repeatedly, its effects often decrease
- 63. Tolerance refers to a slower decrease in response (days or weeks).
- 64. Tolerance may involve increased metabolism of a drug, e.g. ethanol, barbiturates, or homeostatic mechanisms (usually not
- 65. Drug dependence is a term used when a person has a compulsion to take a drug
- 66. Teratogenesis is the occurrence of fetal developmental abnormalities caused by drugs taken during the first trimester
- 68. Carcinogenesis. Drug‐induced tumours are probably very rare because the pharmaceutical industry makes great efforts to avoid
- 70. Скачать презентацию

Medical pharmacology is the science of chemicals (drugs) that interact with
Medical pharmacology is the science of chemicals (drugs) that interact with

Routes of administration: ADVANTAGES and DISADVANTAGES
Oral: the most common and safest,
Routes of administration: ADVANTAGES and DISADVANTAGES
Oral: the most common and safest,
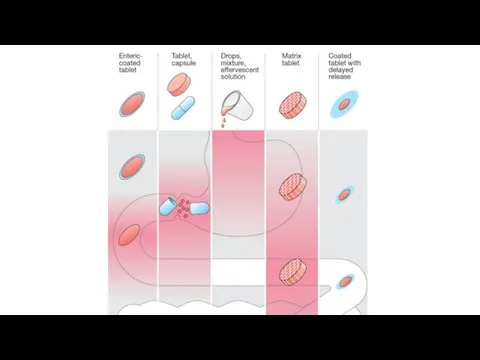

Rectal: partially bypasses first-pass effect, destruction by stomach acid, ideal if
Rectal: partially bypasses first-pass effect, destruction by stomach acid, ideal if

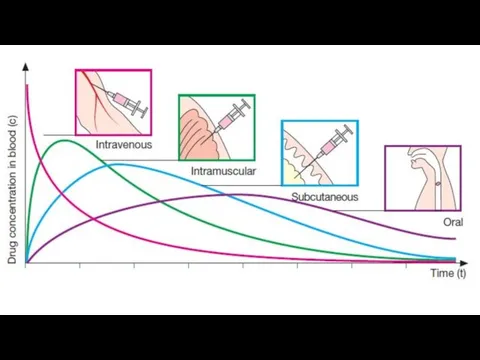
Intravenous: can have immediate effects, ideal if dosed in large volumes,
Intravenous: can have immediate effects, ideal if dosed in large volumes,

Subcutaneous: suitable for slow-release drugs, ideal for some poorly soluble suspensions,
Subcutaneous: suitable for slow-release drugs, ideal for some poorly soluble suspensions,

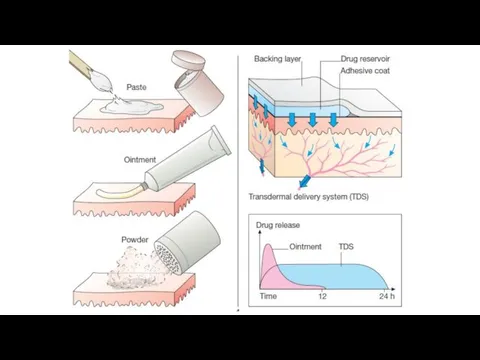
Transdermal (patch): bypasses the first-pass effect, convenient and painless,
ideal
Transdermal (patch): bypasses the first-pass effect, convenient and painless,
ideal

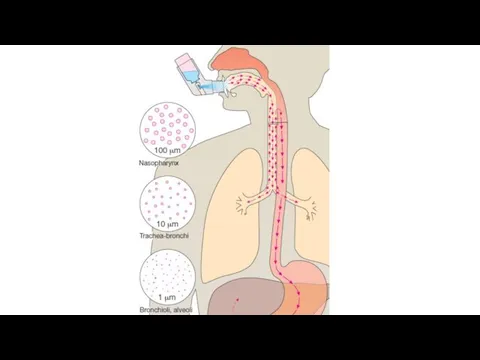
Inhalation: absorption is rapid; can have immediate effects, ideal for gases,
Inhalation: absorption is rapid; can have immediate effects, ideal for gases,

Mechanisms of absorption of drugs
Drugs may be absorbed from the GI
Mechanisms of absorption of drugs
Drugs may be absorbed from the GI

Diffusion: lipid-soluble drugs readily move across most biologic membranes due to
Diffusion: lipid-soluble drugs readily move across most biologic membranes due to

Active transport: Energy-dependent, involves specific carrier proteins. It is capable of
Active transport: Energy-dependent, involves specific carrier proteins. It is capable of
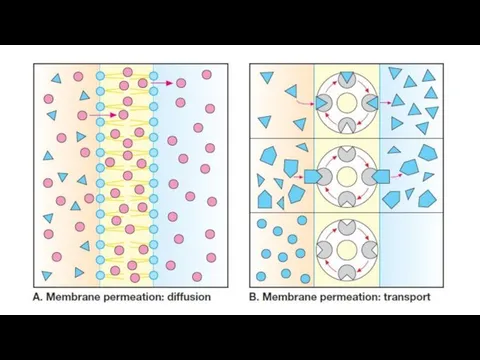
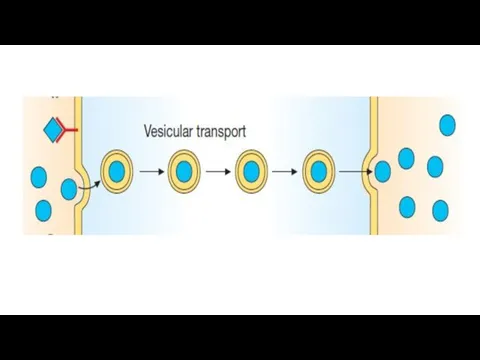

Bioavailability
Bioavailability is the rate and extent to which an administered drug
Bioavailability
Bioavailability is the rate and extent to which an administered drug

Determination of bioavailability: Bioavailability is determined by comparing plasma levels of
Determination of bioavailability: Bioavailability is determined by comparing plasma levels of
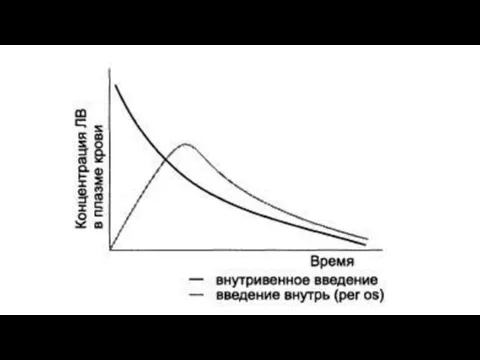

Factors that influence bioavailability
First-pass hepatic metabolism: When a drug is absorbed
Factors that influence bioavailability
First-pass hepatic metabolism: When a drug is absorbed

Chemical instability.
Nature of the drug formulation (salt form, crystal polymorphism, enteric
Chemical instability.
Nature of the drug formulation (salt form, crystal polymorphism, enteric

Drug distribution
Drug distribution is the process by which a drug reversibly
Drug distribution
Drug distribution is the process by which a drug reversibly

Lipid-soluble drugs readily penetrate the CNS because they dissolve in the
Lipid-soluble drugs readily penetrate the CNS because they dissolve in the
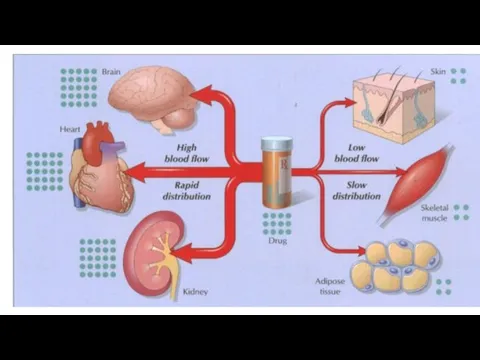
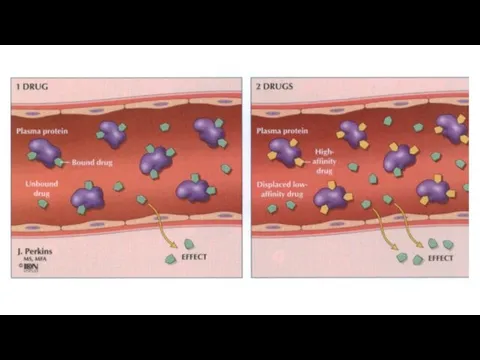

Elimination
Once a drug enters the body, the process of elimination begins.
Elimination
Once a drug enters the body, the process of elimination begins.
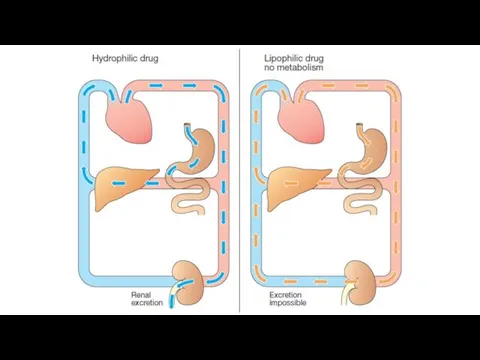

Metabolism in the liver leads to production of products with increased
Metabolism in the liver leads to production of products with increased
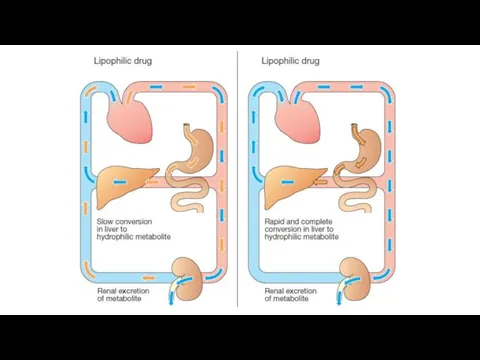

The CYP450-dependent enzymes are an important target for pharmacokinetic drug interactions.
The CYP450-dependent enzymes are an important target for pharmacokinetic drug interactions.

Phase II: This phase consists of conjugation reactions with an endogenous
Phase II: This phase consists of conjugation reactions with an endogenous

Drug clearance may also occur via the intestines, bile, lungs, and
Drug clearance may also occur via the intestines, bile, lungs, and

Pharmacodynamics. Effects
Local effect occurs at the site of drug’s application
Pharmacodynamics. Effects
Local effect occurs at the site of drug’s application

Drugs “’targets
Few drugs (e.g. activated charcoal, osmotic diuretics) act by virtue
Drugs “’targets
Few drugs (e.g. activated charcoal, osmotic diuretics) act by virtue

Enzymes. Drugs that act by inhibiting enzymes include:
anticholinesterases, which enhance
Enzymes. Drugs that act by inhibiting enzymes include:
anticholinesterases, which enhance

Drugs can influence on ion channels (selective pores in the membrane).
Drugs can influence on ion channels (selective pores in the membrane).

However, most drugs produce their effects by acting on specific proteins.
However, most drugs produce their effects by acting on specific proteins.

A receptor as any biologic molecule to which a drug binds
A receptor as any biologic molecule to which a drug binds



3) Kinase‐linked receptors are surface receptors that possess (usually) intrinsic tyrosine
3) Kinase‐linked receptors are surface receptors that possess (usually) intrinsic tyrosine
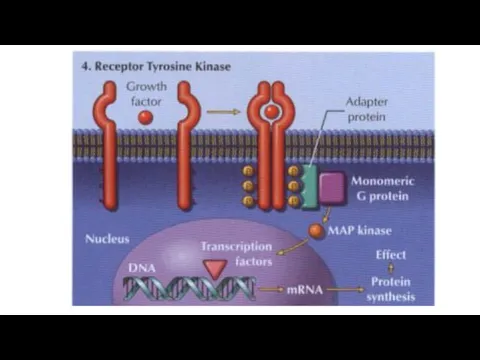
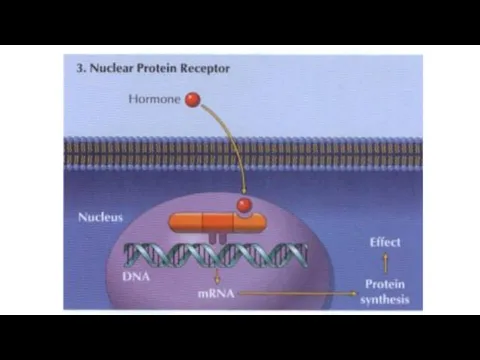

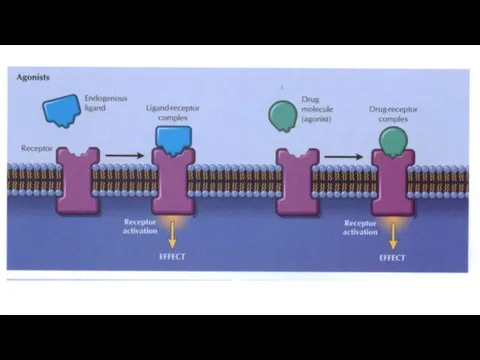
Chemicals (e.g. acetylcholine) or drugs that activate receptors and produce a
Chemicals (e.g. acetylcholine) or drugs that activate receptors and produce a

Partial agonists. These are agonists that cannot elicit the same maximum
Partial agonists. These are agonists that cannot elicit the same maximum

The durability of the “Drug-receptor” bond determines whether the drug action
The durability of the “Drug-receptor” bond determines whether the drug action
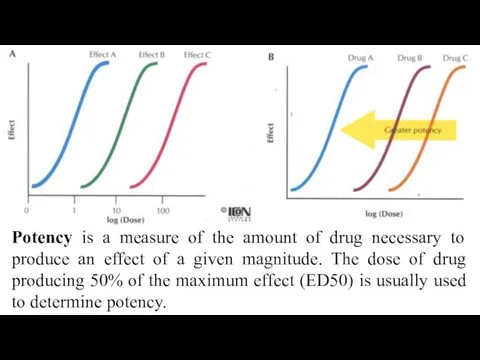
Potency is a measure of the amount of drug necessary to
Potency is a measure of the amount of drug necessary to
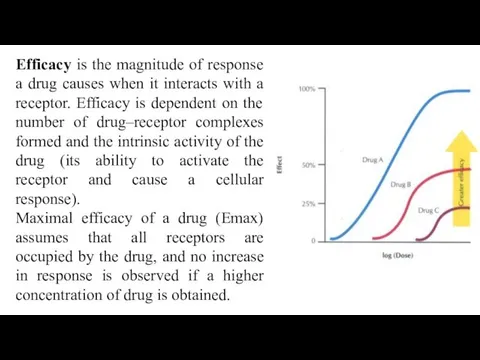
Efficacy is the magnitude of response a drug causes when it
Efficacy is the magnitude of response a drug causes when it

Drugs interactions
Pharmacokinetic
Pharmacodynamic:
Synergism
Antagonism
Drugs interactions
Pharmacokinetic
Pharmacodynamic:
Synergism
Antagonism

In case of synergism drug interaction leads to an increase in
In case of synergism drug interaction leads to an increase in
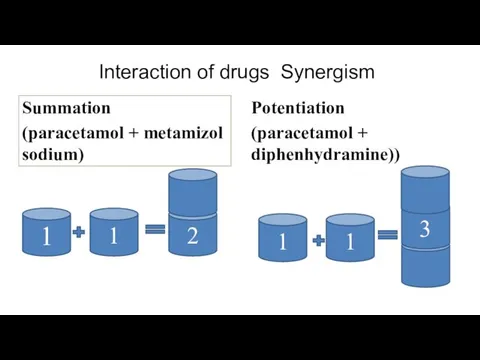
Interaction of drugs Synergism
Summation
(paracetamol + metamizol sodium)
Potentiation
(paracetamol + diphenhydramine))
1
1
2
1
1
3
Interaction of drugs Synergism
Summation
(paracetamol + metamizol sodium)
Potentiation
(paracetamol + diphenhydramine))
1
1
2
1
1
3

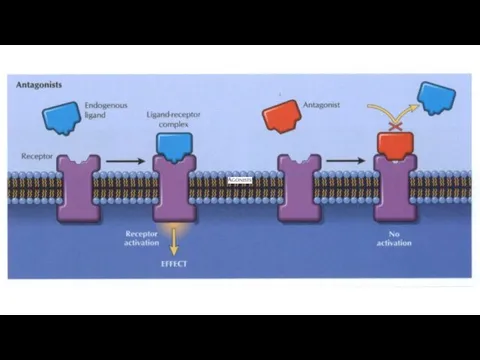
Antagonism
The ability of a drug to decrease the effect of the
Antagonism
The ability of a drug to decrease the effect of the

Irreversible antagonists have an effect that cannot be reversed by increasing
Irreversible antagonists have an effect that cannot be reversed by increasing

Synergoantagonism occurs when some effects of the combined drugs are intensified
Synergoantagonism occurs when some effects of the combined drugs are intensified

Pharmacokinetic interactions
Absorption. Drugs that increase (e.g. metoclopramide) or decrease (e.g. atropine)
Pharmacokinetic interactions
Absorption. Drugs that increase (e.g. metoclopramide) or decrease (e.g. atropine)

Metabolism. Induction of hepatic enzymes by a second drug (e.g.phenobarbital, rifampicin)
Metabolism. Induction of hepatic enzymes by a second drug (e.g.phenobarbital, rifampicin)

Adverse drug reactions
Adverse drug reactions can be divided into those that
Adverse drug reactions
Adverse drug reactions can be divided into those that

Cumulation – storage of pharmacological substance in the body. It is
Cumulation – storage of pharmacological substance in the body. It is
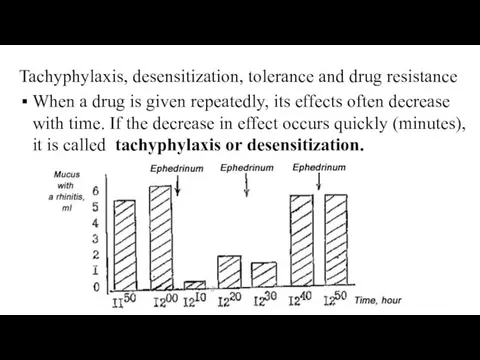
Tachyphylaxis, desensitization, tolerance and drug resistance
When a drug is given repeatedly,
Tachyphylaxis, desensitization, tolerance and drug resistance
When a drug is given repeatedly,

Tolerance refers to a slower decrease in response (days or weeks).
Tolerance refers to a slower decrease in response (days or weeks).

Tolerance may involve increased metabolism of a drug, e.g. ethanol, barbiturates,
Tolerance may involve increased metabolism of a drug, e.g. ethanol, barbiturates,

Drug dependence is a term used when a person has a
Drug dependence is a term used when a person has a

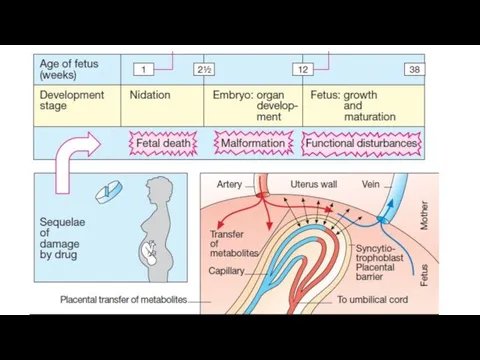
Teratogenesis is the occurrence of fetal developmental abnormalities caused by drugs
Teratogenesis is the occurrence of fetal developmental abnormalities caused by drugs

Carcinogenesis. Drug‐induced tumours are probably very rare because the pharmaceutical industry
Carcinogenesis. Drug‐induced tumours are probably very rare because the pharmaceutical industry
 Сравнительная характеристика состояния полости рта и распространенности заболеваний кариеса между студентами 1-3 курса
Сравнительная характеристика состояния полости рта и распространенности заболеваний кариеса между студентами 1-3 курса Травмы зубов у детей
Травмы зубов у детей Целеполагание в поисковой деятельности
Целеполагание в поисковой деятельности Структура і принципи функціонування імунної системи. Імунологічні методи досліджень. Поняття про імунограму
Структура і принципи функціонування імунної системи. Імунологічні методи досліджень. Поняття про імунограму Абдоминальная хирургия
Абдоминальная хирургия Ультразвуковая (УЗ) терапия
Ультразвуковая (УЗ) терапия Рак яичников
Рак яичников Внимание. Оценка внимания младшего школьника
Внимание. Оценка внимания младшего школьника Инфекционный мононуклеоз
Инфекционный мононуклеоз Здоровый образ жизни и его составляющие
Здоровый образ жизни и его составляющие СДВГ у детей. Неустойчивость, отвлекаемость внимания. Импульсивность. Гиперактивность
СДВГ у детей. Неустойчивость, отвлекаемость внимания. Импульсивность. Гиперактивность Трансформационная игра «Путь к мечте»
Трансформационная игра «Путь к мечте» Межличностные отношения
Межличностные отношения Виды ран и общие правила оказания первой медицинской помощи
Виды ран и общие правила оказания первой медицинской помощи Нейропсихологическое обследование пациентов с деменцией
Нейропсихологическое обследование пациентов с деменцией Коммуникативные свойства личности в теории деятельности
Коммуникативные свойства личности в теории деятельности Other Psychotic Disorders
Other Psychotic Disorders Лучевые реакции и лучевые повреждения
Лучевые реакции и лучевые повреждения Введение в диетологию. Диетотерапия при нарушениях липидного обмена
Введение в диетологию. Диетотерапия при нарушениях липидного обмена Особенности профессиональной деятельности медицинской сестры в профилактике респираторных инфекций
Особенности профессиональной деятельности медицинской сестры в профилактике респираторных инфекций Нагноительные заболевания легких
Нагноительные заболевания легких Американская классическая школа психологии.Теория Г. Эмерсона
Американская классическая школа психологии.Теория Г. Эмерсона Ведение больных после эмболизации маточных сосудов
Ведение больных после эмболизации маточных сосудов Курация рожениц и родильниц
Курация рожениц и родильниц Роль информатизации в системе работы фельдшера медицинской организации
Роль информатизации в системе работы фельдшера медицинской организации Клиническая фармакология обезболевающих лекарственных средств
Клиническая фармакология обезболевающих лекарственных средств Создание композитной накладки своими руками
Создание композитной накладки своими руками Микоплазмоз
Микоплазмоз