- Туа пйда болған және тұқым қуалайтын тубулопатиялар

Содержание
- 2. Тубулопатилар – бұл туа пайда болған немесе жүре пайда болған электролиттер, минералдар, аминоқышқылдар, төменсалмақты ақуыздар ,
- 3. Тубулопатияға қашан күдіктенуге болады? Рахит, Д витаминнің калыпты дозаларына резистентілік Полиурия, полидипсия Өсу және дамудың кідіруі
- 5. Төменгі аяқтардың Рахиттік өзгерістері
- 6. Тубулопатияға күдік туған кездегі негізгі зеріттеулер Зәрдегі және плазмадағы Ca және P Креатинин, Na, K, Cl,
- 7. Нефрондағы заттардың реаборбциясы
- 8. Нефрондағы зат тасымалы
- 9. Нефрондағы зақымдалу деңгейіне байланысты тубулопатияның ерекшеліктері Проксимальді тубулопатиялар Ренальді глюкозурия Фосфат-диабет Аминоацидурия Фанкони синдромы (ДеТони-Дебре) Проксимальді
- 10. Ренальді глюкозурия Аутосомды-доминантты тұқым куалау (А типі) Глюкозаның бүйректік табалдырығының төмендеуі. Глюказының зәрмен гликемиясыз жоғалуы 2-30
- 11. Аминоацидурия Нейтральді аминоацидурия Хартнупа ауруы – 12 аминоқышқылдың реобсорбциясының бұзылысы сонымен катар Триптофан бұзылысымен ақыл есінің
- 12. Негізгі аминоацидурия Цистинурия - а/р тұқым қуалау, жиілігі 1:7000 нәрестелер Цистин, лизин, аргинин, орнитиннің апикальді тасмалдануының
- 13. Негізгі аминоацидурия Лизинуриялық протеинуриялық көтереалмаушылық – зәр қышқылы айналымының бұзылысы мен гипераммонемия қатар жүретін жасушаішілік концентрациясының
- 14. Кальцидің нефрондағы тасымалдануы
- 15. Фанкони синдромы Симптомдары: * дамуының кешеуілдеуі * рахит * полиурия Лабораторлы: * ацидоз * гипокалиемия *
- 16. Фанкони синдромымен туа пайда болған аурулар
- 17. Фанкони синдромымен туа пайда болған аурулар-2
- 18. Фанкони синдромымен туа пайда болған аурулар-3
- 19. Фанкони синдромы емі Спецификалық, ФС корригирлеуге болады Сілтілі ерітінеділер(Albright, Lightwood или Sohlґs ) K алмастыру P
- 20. Фосфат-диабеті – Х-қосарланған гипофосфатемикалық рахит Доминантты немесе рецессивті тұқымқуалау-1:20 000 нәресте Форсфаттардың реабсорбциясының проксимальды ақауы Бұған
- 21. Дента аруы Дента ауруы - Х-қосарланған рецессивті нефролитиаз 746 аминқышқылдарынан протеин кодтаушы Хр11.22 хромасомасында CLCN5 бүйректік
- 22. Клинические проявления болезни Дента Гиперкальцийурия Медуллярный нефрокальциноз/нефролитиаз Низкомолекулярная протеинурия Прогрессирующее снижение почечных функций Аминоацидурия +/- Фосфатурия
- 23. Нефрокальциноз при болезни Дента
- 24. Синдром Барттера – гипокалиемический, гипохлоремичечкий метаболический алкалоз Мутация : * Na-K-2Cl транспортера * K-канала * Cl-канала
- 25. Синдром Гительмана Дефект тиазид чувствительного ко-транспортера Na-Cl, аутосомно рецессивн Симптомы: * низкий K, алкалоз * низкий
- 26. Виды дистального ренального тубулярного ацидоза ( I типа) I ПЕРВИЧНЫЙ - наследственный (аутосомно-рецессивный с или без
- 27. Виды дистального ренального тубулярного ацидоза ( I типа) Гиперкальциурия и нефрокальциноз - первичный гиперпаратиреоидизм - интоксикация
- 28. Клиника ДРТА у детей Манифестация в возрасте 3 мес-2 лет Задержка роста и прибавки массы тела
- 29. Схема патогенеза ДРТА Нарушение экскреции Н Ацидоз Снижение экскреции цитрата гиперкальцийурия Мобилизация солей Са из костей
- 30. Диагностика первичного ДРТА Метаболический гиперхлоремический ацидоз с нормальными показателями анионной разницы плазмы ( Na-Cl-HCO3 = 8-16
- 31. Нефрокальциноз при семейном дистальном РТА
- 32. Принципы лечения ДРТА Коррекция ацидоза ( бикарбонат натрия в дозе 1-3 ммоль\кг у детей старше 5
- 33. Псевдогипоальдостеронизм Аутосомно рецессивный, дефект рецепторов к минералкортикоидам Симптомы: * низкий Na, высокий K * повышена экскреция
- 34. Синдром Liddle Аутосомно-доминантное заболевание Гипертензия Гипокалиемический метаболический алкалоз Снижение уровней ренина и альдостерона Повышение реабсорбции натрия
- 35. Транспорт воды в собирательных трубочках нефрона
- 36. Нефрогенный несахарный диабет X-сцеплен рецессив., мутация гена рецептора V2R Аутосомно рецесс., мутация гена рецептора AQP2 Симптомы:
- 38. Скачать презентацию

Тубулопатилар – бұл туа пайда болған немесе жүре пайда болған электролиттер,
Тубулопатилар – бұл туа пайда болған немесе жүре пайда болған электролиттер,

Тубулопатияға қашан күдіктенуге болады?
Рахит, Д витаминнің калыпты дозаларына резистентілік
Полиурия, полидипсия
Өсу
Тубулопатияға қашан күдіктенуге болады?
Рахит, Д витаминнің калыпты дозаларына резистентілік
Полиурия, полидипсия
Өсу

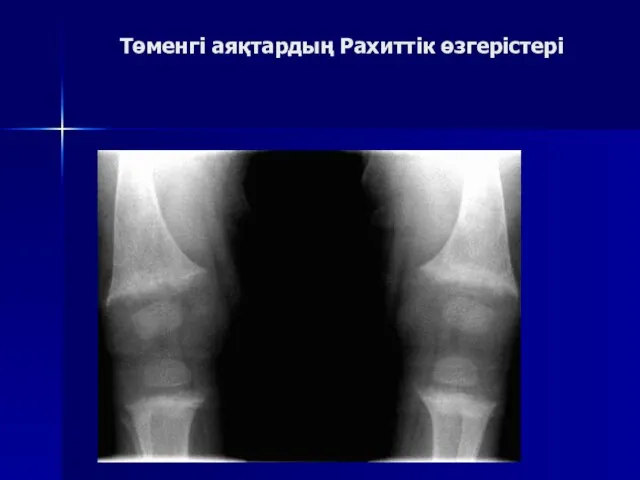
Төменгі аяқтардың Рахиттік өзгерістері
Төменгі аяқтардың Рахиттік өзгерістері

Тубулопатияға күдік туған кездегі негізгі зеріттеулер
Зәрдегі және плазмадағы Ca және P
Креатинин,
Тубулопатияға күдік туған кездегі негізгі зеріттеулер
Зәрдегі және плазмадағы Ca және P
Креатинин,
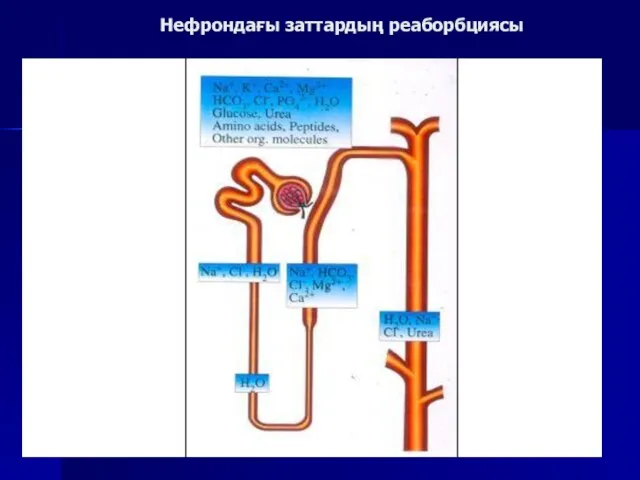
Нефрондағы заттардың реаборбциясы
Нефрондағы заттардың реаборбциясы
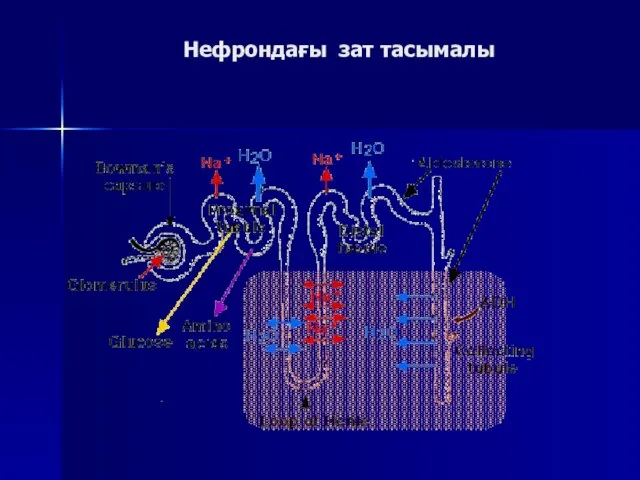
Нефрондағы зат тасымалы
Нефрондағы зат тасымалы

Нефрондағы зақымдалу деңгейіне байланысты тубулопатияның ерекшеліктері
Проксимальді тубулопатиялар
Ренальді глюкозурия
Фосфат-диабет
Аминоацидурия
Фанкони синдромы (ДеТони-Дебре)
Проксимальді РТА
Нефрондағы зақымдалу деңгейіне байланысты тубулопатияның ерекшеліктері
Проксимальді тубулопатиялар
Ренальді глюкозурия
Фосфат-диабет
Аминоацидурия
Фанкони синдромы (ДеТони-Дебре)
Проксимальді РТА

Ренальді глюкозурия
Аутосомды-доминантты тұқым куалау (А типі)
Глюкозаның бүйректік табалдырығының төмендеуі.
Глюказының зәрмен гликемиясыз
Ренальді глюкозурия
Аутосомды-доминантты тұқым куалау (А типі)
Глюкозаның бүйректік табалдырығының төмендеуі.
Глюказының зәрмен гликемиясыз

Аминоацидурия
Нейтральді аминоацидурия
Хартнупа ауруы – 12 аминоқышқылдың реобсорбциясының бұзылысы сонымен катар
Аминоацидурия
Нейтральді аминоацидурия
Хартнупа ауруы – 12 аминоқышқылдың реобсорбциясының бұзылысы сонымен катар

Негізгі аминоацидурия
Цистинурия - а/р тұқым қуалау, жиілігі 1:7000 нәрестелер
Цистин, лизин, аргинин,
Негізгі аминоацидурия
Цистинурия - а/р тұқым қуалау, жиілігі 1:7000 нәрестелер
Цистин, лизин, аргинин,

Негізгі аминоацидурия
Лизинуриялық протеинуриялық көтереалмаушылық – зәр қышқылы айналымының бұзылысы мен гипераммонемия
Негізгі аминоацидурия
Лизинуриялық протеинуриялық көтереалмаушылық – зәр қышқылы айналымының бұзылысы мен гипераммонемия

Кальцидің нефрондағы тасымалдануы
Кальцидің нефрондағы тасымалдануы

Фанкони синдромы
Симптомдары: * дамуының кешеуілдеуі
* рахит
* полиурия
Лабораторлы: *
Фанкони синдромы
Симптомдары: * дамуының кешеуілдеуі
* рахит
* полиурия
Лабораторлы: *

Фанкони синдромымен туа пайда болған аурулар
Фанкони синдромымен туа пайда болған аурулар
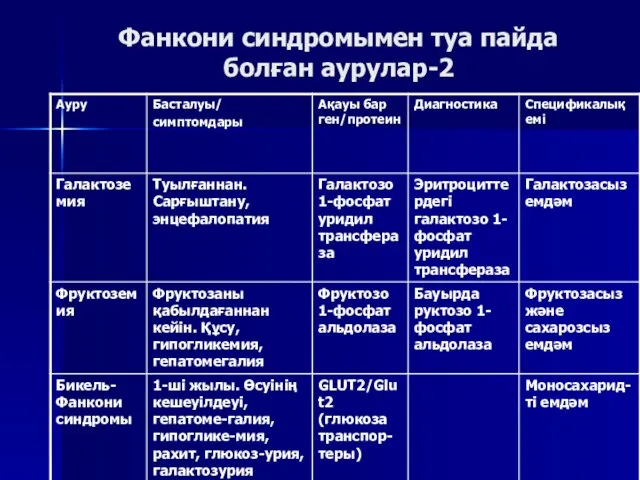
Фанкони синдромымен туа пайда болған аурулар-2
Фанкони синдромымен туа пайда болған аурулар-2

Фанкони синдромымен туа пайда болған аурулар-3
Фанкони синдромымен туа пайда болған аурулар-3

Фанкони синдромы емі
Спецификалық, ФС корригирлеуге болады
Сілтілі ерітінеділер(Albright, Lightwood или Sohlґs )
K
Фанкони синдромы емі
Спецификалық, ФС корригирлеуге болады
Сілтілі ерітінеділер(Albright, Lightwood или Sohlґs )
K

Фосфат-диабеті – Х-қосарланған гипофосфатемикалық рахит
Доминантты немесе рецессивті тұқымқуалау-1:20 000 нәресте
Форсфаттардың реабсорбциясының
Фосфат-диабеті – Х-қосарланған гипофосфатемикалық рахит
Доминантты немесе рецессивті тұқымқуалау-1:20 000 нәресте
Форсфаттардың реабсорбциясының

Дента аруы
Дента ауруы - Х-қосарланған рецессивті нефролитиаз
746 аминқышқылдарынан протеин кодтаушы
Дента аруы
Дента ауруы - Х-қосарланған рецессивті нефролитиаз
746 аминқышқылдарынан протеин кодтаушы

Клинические проявления болезни Дента
Гиперкальцийурия
Медуллярный нефрокальциноз/нефролитиаз
Низкомолекулярная протеинурия
Прогрессирующее снижение почечных функций
Аминоацидурия +/-
Фосфатурия +/-
Глюкозурия
Клинические проявления болезни Дента
Гиперкальцийурия
Медуллярный нефрокальциноз/нефролитиаз
Низкомолекулярная протеинурия
Прогрессирующее снижение почечных функций
Аминоацидурия +/-
Фосфатурия +/-
Глюкозурия

Нефрокальциноз
при болезни Дента
Нефрокальциноз
при болезни Дента

Синдром Барттера – гипокалиемический, гипохлоремичечкий метаболический алкалоз
Мутация : * Na-K-2Cl транспортера
Синдром Барттера – гипокалиемический, гипохлоремичечкий метаболический алкалоз
Мутация : * Na-K-2Cl транспортера

Синдром Гительмана
Дефект тиазид чувствительного ко-транспортера Na-Cl, аутосомно рецессивн
Симптомы: * низкий K,
Синдром Гительмана
Дефект тиазид чувствительного ко-транспортера Na-Cl, аутосомно рецессивн
Симптомы: * низкий K,

Виды дистального ренального тубулярного ацидоза ( I типа)
I ПЕРВИЧНЫЙ
- наследственный
Виды дистального ренального тубулярного ацидоза ( I типа)
I ПЕРВИЧНЫЙ
- наследственный

Виды дистального ренального тубулярного ацидоза ( I типа)
Гиперкальциурия и нефрокальциноз
-
Виды дистального ренального тубулярного ацидоза ( I типа)
Гиперкальциурия и нефрокальциноз
-

Клиника ДРТА у детей
Манифестация в возрасте 3 мес-2 лет
Задержка роста и
Клиника ДРТА у детей
Манифестация в возрасте 3 мес-2 лет
Задержка роста и
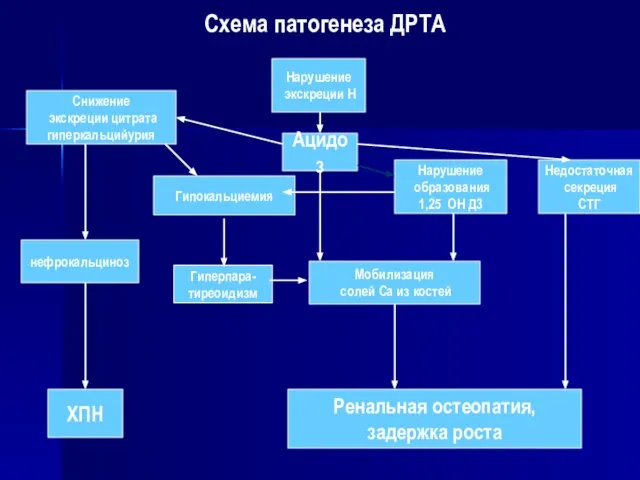
Схема патогенеза ДРТА
Нарушение
экскреции Н
Ацидоз
Снижение
экскреции цитрата
гиперкальцийурия
Мобилизация
солей Са из костей
Нарушение
Схема патогенеза ДРТА
Нарушение
экскреции Н
Ацидоз
Снижение
экскреции цитрата
гиперкальцийурия
Мобилизация
солей Са из костей
Нарушение

Диагностика первичного ДРТА
Метаболический гиперхлоремический ацидоз с нормальными показателями анионной разницы плазмы
Диагностика первичного ДРТА
Метаболический гиперхлоремический ацидоз с нормальными показателями анионной разницы плазмы
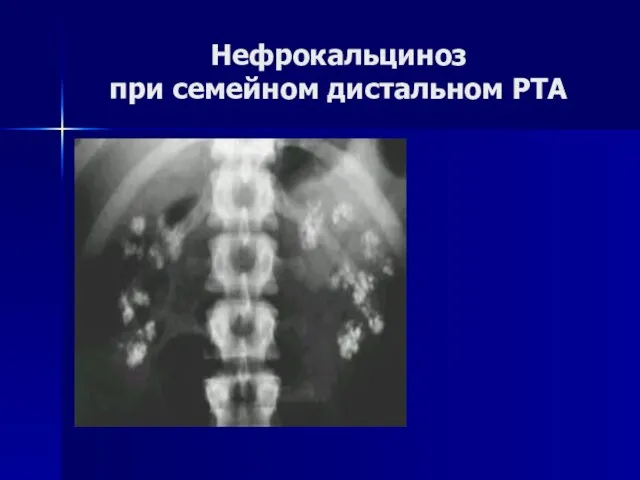
Нефрокальциноз
при семейном дистальном РТА
Нефрокальциноз
при семейном дистальном РТА

Принципы лечения ДРТА
Коррекция ацидоза ( бикарбонат натрия в дозе 1-3
Принципы лечения ДРТА
Коррекция ацидоза ( бикарбонат натрия в дозе 1-3

Псевдогипоальдостеронизм
Аутосомно рецессивный, дефект рецепторов к минералкортикоидам
Симптомы: * низкий Na, высокий K
Псевдогипоальдостеронизм
Аутосомно рецессивный, дефект рецепторов к минералкортикоидам
Симптомы: * низкий Na, высокий K

Синдром Liddle
Аутосомно-доминантное заболевание
Гипертензия
Гипокалиемический метаболический алкалоз
Снижение уровней ренина и альдостерона
Повышение реабсорбции натрия
Синдром Liddle
Аутосомно-доминантное заболевание
Гипертензия
Гипокалиемический метаболический алкалоз
Снижение уровней ренина и альдостерона
Повышение реабсорбции натрия
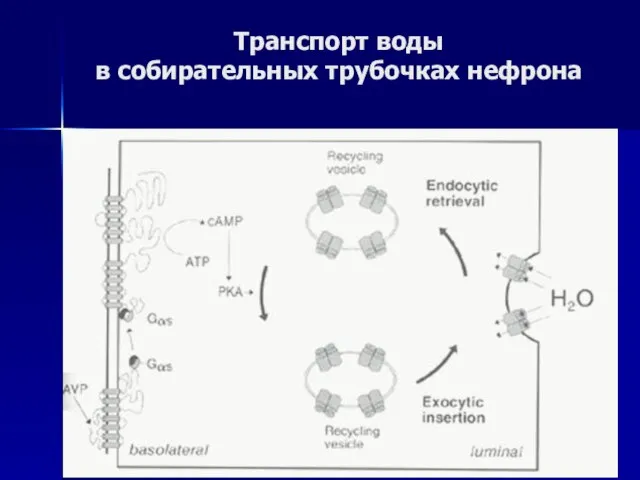
Транспорт воды
в собирательных трубочках нефрона
Транспорт воды
в собирательных трубочках нефрона

Нефрогенный несахарный диабет
X-сцеплен рецессив., мутация гена рецептора V2R
Аутосомно рецесс., мутация
Нефрогенный несахарный диабет
X-сцеплен рецессив., мутация гена рецептора V2R
Аутосомно рецесс., мутация
 Организация первой помощи пострадавшим
Организация первой помощи пострадавшим Оказание первой помощи при несчастных случаях на производстве
Оказание первой помощи при несчастных случаях на производстве Одонтогенные опухоли и эпителиальные кисты челюстей
Одонтогенные опухоли и эпителиальные кисты челюстей Сальмонеллёзы
Сальмонеллёзы Острый холецистит
Острый холецистит Лихорадочный синдром и субфебрилитет
Лихорадочный синдром и субфебрилитет Ортобиотическая система комплексной реабилитации и социальной адаптации детей с тяжёлыми нарушениями речи
Ортобиотическая система комплексной реабилитации и социальной адаптации детей с тяжёлыми нарушениями речи Das Gefäßzentrum Niederlausitz Igor Grobel 20. Symposium der Vereinigung der Gefäßchirurgen Brandenburgs Bad Saarow
Das Gefäßzentrum Niederlausitz Igor Grobel 20. Symposium der Vereinigung der Gefäßchirurgen Brandenburgs Bad Saarow Реставрация костного препарата (черепа)
Реставрация костного препарата (черепа) Влияние веса школьного портфеля на здоровье школьника
Влияние веса школьного портфеля на здоровье школьника Промышленные яды и их классификация. Показатели токсичности. Общие закономерности действия и направления профилактики
Промышленные яды и их классификация. Показатели токсичности. Общие закономерности действия и направления профилактики Первая медицинская помощь при травмах: порезах, ссадинах, ушибах, вывихах, переломах
Первая медицинская помощь при травмах: порезах, ссадинах, ушибах, вывихах, переломах Історія хвороби: розсіяний склероз
Історія хвороби: розсіяний склероз Наследственные болезни и их классификация. Генные болезни
Наследственные болезни и их классификация. Генные болезни Этикет в деловой коммуникации
Этикет в деловой коммуникации Участие в работе перевязочного кабинета хирургического отделения
Участие в работе перевязочного кабинета хирургического отделения Детский оздоровительный лагерь Дон
Детский оздоровительный лагерь Дон Зоонозные заболевания
Зоонозные заболевания Генерализованная туляремия. Клинический случай
Генерализованная туляремия. Клинический случай Профилактика передачи ВИЧ-инфекции в работе медицинской сестры
Профилактика передачи ВИЧ-инфекции в работе медицинской сестры Соединение костей. Лекция №13
Соединение костей. Лекция №13 Сахарный диабет. Практическое занятие
Сахарный диабет. Практическое занятие Акушерские кровотечения в родах и послеродовом периоде. Тема 3
Акушерские кровотечения в родах и послеродовом периоде. Тема 3 Препараты гормонов и их синтетические заменители
Препараты гормонов и их синтетические заменители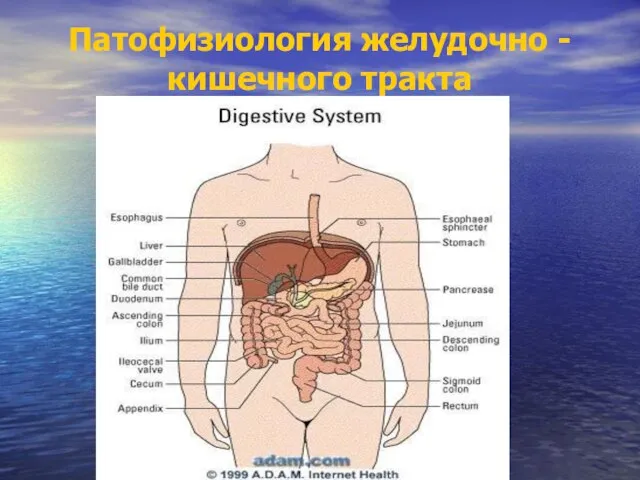 Патофизиология желудочно - кишечного тракта
Патофизиология желудочно - кишечного тракта Коматозные состояния в практике невролога
Коматозные состояния в практике невролога Травматизм. Виды травматизма
Травматизм. Виды травматизма Заболевания органов пищеварения
Заболевания органов пищеварения