- Моделирование, как метод научного исследования. Ограниченный метод Хартри-Фока

Содержание
- 2. СОДЕРЖАНИЕ 1. Моделирование как метод научного исследования 2. Ограниченный метод Хартри – Фока
- 4. Ду́глас Ре́йнер Ха́ртри (англ. Douglas Rayner Hartree; 27 марта 1897, Кембридж — 12 февраля 1958, Кембридж)
- 5. Влади́мир Алекса́ндрович Фок (1898—1974) — советский физик. Академик АН СССР (1939; член-корреспондент с 1932 года). Герой
- 6. Метод Хартри – Фока является основным расчетным методом квантовой химии. Он представлен в различных вариантах: -для
- 7. Метод широко используется в квантовой химии, в частности, для проведения численного моделирования конфигурации некоторых молекул, в
- 8. ПОЛУЭМПИРИЧЕСКИЕ МЕТОДЫ квантовой химии, методы расчета молекулярных характеристик или свойств вещества с привлечением экспериментальных данных. По
- 9. Метод INDO занимает промежуточное по сложности и времени вычислений положение между методами NDDO и CNDO. Он
- 10. Метод NDDO. На данном приближении основаны наиболее точные полуэмпирические методы, наибольшее распространение получили процедуры MNDO (М.
- 11. Метод MNDO. В методе MNDO(Modified Neglect of Diatomic Overlap, МПДП) учитываются интегралы межэлектронного отталкивания, включающие одноцентровые
- 12. Метод AM1. С целью учета этих эффектов в параметризации AM1 (Austin Model) в выражении для Ecore
- 13. Метод PM3. Метод PM3 очень близок к методу AM1, отличие состоит в том, что в методе
- 15. Скачать презентацию

СОДЕРЖАНИЕ
1. Моделирование как метод научного исследования
2. Ограниченный метод Хартри –
СОДЕРЖАНИЕ
1. Моделирование как метод научного исследования 2. Ограниченный метод Хартри –


Ду́глас Ре́йнер Ха́ртри (англ. Douglas Rayner Hartree; 27 марта 1897, Кембридж — 12 февраля 1958, Кембридж) — английский физик-теоретик и математик. Член Лондонского
Ду́глас Ре́йнер Ха́ртри (англ. Douglas Rayner Hartree; 27 марта 1897, Кембридж — 12 февраля 1958, Кембридж) — английский физик-теоретик и математик. Член Лондонского

Влади́мир Алекса́ндрович Фок (1898—1974) — советский физик. Академик АН СССР (1939; член-корреспондент с 1932 года). Герой Социалистического Труда.
Член ряда академий наук
Влади́мир Алекса́ндрович Фок (1898—1974) — советский физик. Академик АН СССР (1939; член-корреспондент с 1932 года). Герой Социалистического Труда. Член ряда академий наук

Метод Хартри – Фока является основным расчетным методом квантовой химии.
Он
Метод Хартри – Фока является основным расчетным методом квантовой химии.
Он

Метод широко используется в квантовой химии, в частности, для проведения численного моделирования конфигурации
Метод широко используется в квантовой химии, в частности, для проведения численного моделирования конфигурации

ПОЛУЭМПИРИЧЕСКИЕ МЕТОДЫ квантовой химии, методы расчета молекулярных характеристик или свойств вещества с
ПОЛУЭМПИРИЧЕСКИЕ МЕТОДЫ квантовой химии, методы расчета молекулярных характеристик или свойств вещества с

Метод INDO занимает промежуточное по сложности и времени вычислений положение между
Метод INDO занимает промежуточное по сложности и времени вычислений положение между

Метод NDDO. На данном приближении основаны наиболее точные полуэмпирические методы, наибольшее
Метод NDDO. На данном приближении основаны наиболее точные полуэмпирические методы, наибольшее

Метод MNDO. В методе MNDO(Modified Neglect of Diatomic Overlap, МПДП) учитываются
Метод MNDO. В методе MNDO(Modified Neglect of Diatomic Overlap, МПДП) учитываются

Метод AM1. С целью учета этих эффектов в параметризации AM1 (Austin
Метод AM1. С целью учета этих эффектов в параметризации AM1 (Austin

Метод PM3. Метод PM3 очень близок к методу AM1, отличие состоит
Метод PM3. Метод PM3 очень близок к методу AM1, отличие состоит
 Типы химических реакций
Типы химических реакций Электролиз. 9 класс
Электролиз. 9 класс Кристаллооптический метод в петрографии
Кристаллооптический метод в петрографии Организация дипептида
Организация дипептида Реакция Пфитцингера
Реакция Пфитцингера Презентация Строение газообразных, жидких и твёрдых тел
Презентация Строение газообразных, жидких и твёрдых тел Роль воды в жизнедеятельности организма. Теория растворов электролитов и неэлектролитов коллигативные свойства растворов
Роль воды в жизнедеятельности организма. Теория растворов электролитов и неэлектролитов коллигативные свойства растворов Нуклеиновые кислоты
Нуклеиновые кислоты Илік заттар (немесе тұтқыр заттар), тітіркендіруші заттар.қаптаушы заттар, адсорбциялаушы заттар
Илік заттар (немесе тұтқыр заттар), тітіркендіруші заттар.қаптаушы заттар, адсорбциялаушы заттар Металловедение. Материалы для изготовления металлоконструкций. (Лекция 7)
Металловедение. Материалы для изготовления металлоконструкций. (Лекция 7) Генетическая связь между классами веществ
Генетическая связь между классами веществ Кислотно-основные взаимодействия. Принцип ЖМКО Пирсона
Кислотно-основные взаимодействия. Принцип ЖМКО Пирсона Экологический проект: Определение химического состава местных глин
Экологический проект: Определение химического состава местных глин Проверьте себя: Электроотрицательность - это ……. Ковалентная полярная связь – это ….. Ковалентная неполярная связь – это ….. Элемент, имеющий самое высокое значение электроотрицательности ……. Какую кристаллическую решетку имеют вещества с ковалентны
Проверьте себя: Электроотрицательность - это ……. Ковалентная полярная связь – это ….. Ковалентная неполярная связь – это ….. Элемент, имеющий самое высокое значение электроотрицательности ……. Какую кристаллическую решетку имеют вещества с ковалентны Бензол. Получение бензола. Химические свойства бензола. Применение бензола на основе его свойств
Бензол. Получение бензола. Химические свойства бензола. Применение бензола на основе его свойств Гетероциклдік қосылыстар
Гетероциклдік қосылыстар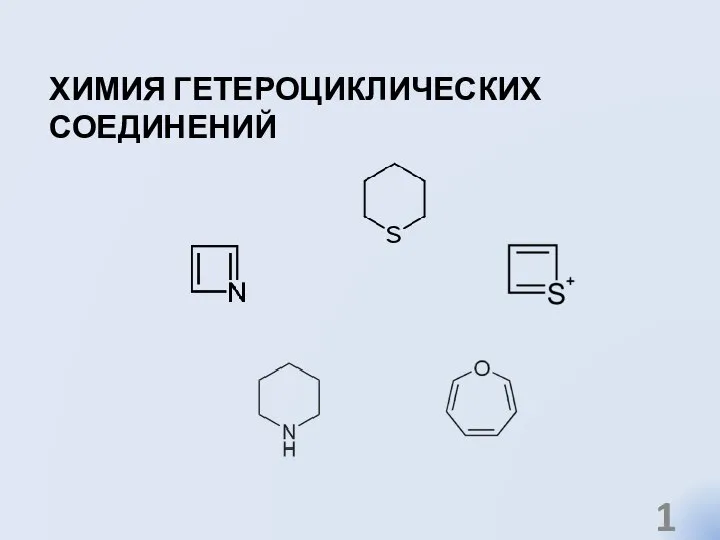 Химия гетероциклических соединений
Химия гетероциклических соединений Депонирование и мобилизация жиров
Депонирование и мобилизация жиров Натуральный каучук
Натуральный каучук Химический источник электрического тока
Химический источник электрического тока Анализ физико-химических свойств синтетических моющих средств (СМС) и их значение в жизни человека
Анализ физико-химических свойств синтетических моющих средств (СМС) и их значение в жизни человека Курсовая работа: синтез n - бензил – n – этилэтанамина (диэтилбензиламина)
Курсовая работа: синтез n - бензил – n – этилэтанамина (диэтилбензиламина) Трансляция. Активирование аминокислоты
Трансляция. Активирование аминокислоты Виды химической связи (8 класс)
Виды химической связи (8 класс) ГИА-9 Химия. А4
ГИА-9 Химия. А4 Автометаморфизм
Автометаморфизм Изотермический процесс в реакционном объеме. (Тема 6.2)
Изотермический процесс в реакционном объеме. (Тема 6.2) МУНИЦИПАЛЬНОЕ ОБЩЕОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ «СРЕДНЯЯ ОБЩЕОБРАЗОВАТЕЛЬНАЯ ШКОЛА №22»
МУНИЦИПАЛЬНОЕ ОБЩЕОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ «СРЕДНЯЯ ОБЩЕОБРАЗОВАТЕЛЬНАЯ ШКОЛА №22»