- Болезнь Фабри

Содержание
- 2. ОПРЕДЕЛЕНИЕ Болезнь Фабри или болезнь Андерсона-Фабри – наследственное заболевание, относящееся к группе лизосомных болезней накопления, обусловленное
- 3. ИСТОРИЧЕСКАЯ СПРАВКА Болезнь названа в честь одного из его первооткрывателей — Джона Фабри (1 июня 1860
- 4. ЭПИДЕМИОЛОГИЯ Болезнь Фабри относится к редким заболеваниям. Распространенность болезни в различных странах мира варьирует в широких
- 5. ЭТИОЛОГИЯ Причиной возникновения болезни Фабри являются мутации гена GLA, контролирующего структуру α-галактозидазы А (GLA; EC 3.2.1.22).
- 6. Болезнь Фабри наследуется по рецессивному X-сцепленному типу. Гемизиготные мужчины имеют единственную мутантную Х-хромосому, что определяет классический
- 7. ПАТОГЕНЕЗ Первичным патогенетическим звеном болезнь Фабри является дефицит α-галактозидазы А. которая в норме отщепляет терминальный остаток
- 8. КЛАССИФИКАЦИЯ Выделяют две формы болезни: Классическая Атипичная (позднее начало, изолированное поражение головного мозга, сердца или почек).
- 9. КЛИНИЧЕСКАЯ КАРТИНА Клинические фенотипы при болезни Фабри могут быть чрезвычайно разнообразными как у лиц мужского, так
- 10. Кожные проявления. Ангиокератомы состоят из скоплений отдельных эктатических кровеносных сосудов, покрытых несколькими слоями кожи. Обычно расположены
- 11. Поражение почек. Первые симптомы – микроальбуминурия и протеинурия появляются в возрасте 20-30 лет. С возрастом протеинурия
- 12. Поражение сердца. Поражение сердца относят к числу распространенных и прогностически неблагоприятных проявлений болезни Фабри. Именно заболевания
- 13. Офтальмологические нарушения. Типичным симптомом болезни Фабри является помутнение роговицы в виде завитков (70-90% больных). Наблюдается задняя
- 14. Изменения со стороны органов слуха и вестибулярного аппарата. У большинства пациентов с болезнью Фабри отмечается прогрессирующее
- 15. Периферическая нервная система. Болезнь Фабри относится к болезненным полиневропатиям. У пациентов с болезнью Фабри в 70–80%
- 16. Для болезни Фабри типичны болевые кризы, которые часто возникают при перемене погоды, лихорадке, физической нагрузке, стрессе
- 17. Центральная нервная система. Болезнь Фабри можно отнести к болезни малых сосудов (малых артерий и артериол). КЛИНИЧЕСКАЯ
- 18. У большинства больных инсульт развивается в возрасте от 20 до 50 лет, в том числе у
- 19. ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА Заподозрить болезнь Фабри проще при наличии классического варианта – системного заболевания с поражением почек,
- 20. ДНК - ДИАГНОСТИКА случае неоднозначных результатов биохимических исследований у больных, а также с целью проведения пренатальной
- 21. ДИАГНОСТИКА Диагностика болезни Фабри включает комплексную оценку клинический картины, лабораторные тесты и другие исследования (например, исследование
- 22. ЛЕЧЕНИЕ ИСПОЛЬЗОВАНИЕ ФЕРМЕНТОЗАМЕСТИТЕЛЬНОЙ ТЕРАПИИ (ФЗТ). Заместительная терапия рекомбинантными препаратами альфа-галактозидазы А используется с 2001 года. С
- 23. СИМПТОМАТИЧЕСКАЯ ТЕРАПИЯ У пациентов с хронической невропатической болью и акропарестезиями целесообразно по возможности устранить факторы, провоцирующие
- 24. У пациентов с диспепсией возможно лечение блокаторами H2-гистаминовых рецепторов или ингибиторами протонной помпы. Для уменьшения протеинурии
- 25. ПРОГНОЗ При своевременном доступе к ФЗТ, надлежащем мониторинге болезни и соблюдении врачебных рекомендаций прогноз благоприятный.
- 27. Скачать презентацию
ОПРЕДЕЛЕНИЕ
Болезнь Фабри или болезнь Андерсона-Фабри – наследственное заболевание, относящееся к
ОПРЕДЕЛЕНИЕ
Болезнь Фабри или болезнь Андерсона-Фабри – наследственное заболевание, относящееся к

ИСТОРИЧЕСКАЯ СПРАВКА
Болезнь названа в честь одного из его первооткрывателей — Джона Фабри
ИСТОРИЧЕСКАЯ СПРАВКА
Болезнь названа в честь одного из его первооткрывателей — Джона Фабри

ЭПИДЕМИОЛОГИЯ
Болезнь Фабри относится к редким заболеваниям.
Распространенность болезни в различных
ЭПИДЕМИОЛОГИЯ
Болезнь Фабри относится к редким заболеваниям.
Распространенность болезни в различных

ЭТИОЛОГИЯ
Причиной возникновения болезни Фабри являются мутации гена GLA, контролирующего структуру
ЭТИОЛОГИЯ
Причиной возникновения болезни Фабри являются мутации гена GLA, контролирующего структуру
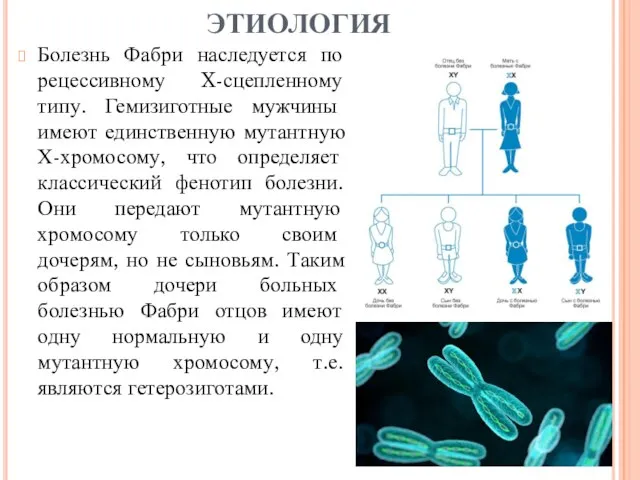
Болезнь Фабри наследуется по рецессивному X-сцепленному типу. Гемизиготные мужчины имеют единственную
Болезнь Фабри наследуется по рецессивному X-сцепленному типу. Гемизиготные мужчины имеют единственную
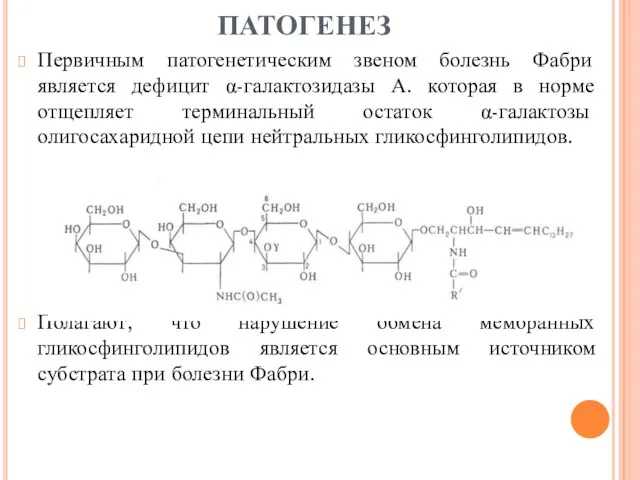
ПАТОГЕНЕЗ
Первичным патогенетическим звеном болезнь Фабри является дефицит α-галактозидазы А. которая в
ПАТОГЕНЕЗ
Первичным патогенетическим звеном болезнь Фабри является дефицит α-галактозидазы А. которая в
КЛАССИФИКАЦИЯ
Выделяют две формы болезни:
Классическая
Атипичная
(позднее начало, изолированное поражение головного мозга, сердца
КЛАССИФИКАЦИЯ
Выделяют две формы болезни:
Классическая
Атипичная
(позднее начало, изолированное поражение головного мозга, сердца

КЛИНИЧЕСКАЯ КАРТИНА
Клинические фенотипы при болезни Фабри могут быть чрезвычайно разнообразными как
КЛИНИЧЕСКАЯ КАРТИНА
Клинические фенотипы при болезни Фабри могут быть чрезвычайно разнообразными как
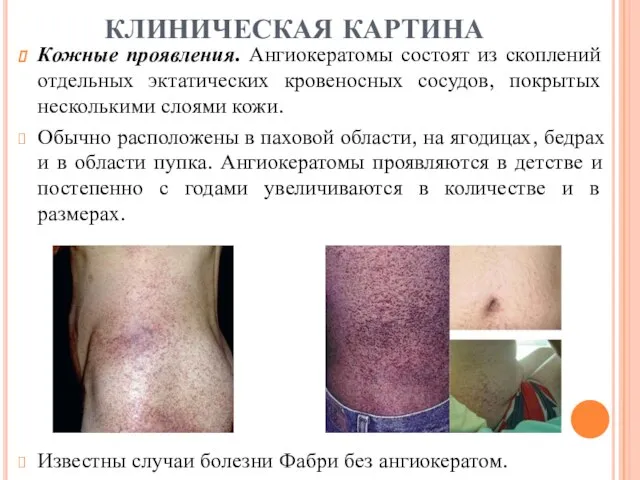
Кожные проявления. Ангиокератомы состоят из скоплений отдельных эктатических кровеносных сосудов, покрытых
Кожные проявления. Ангиокератомы состоят из скоплений отдельных эктатических кровеносных сосудов, покрытых
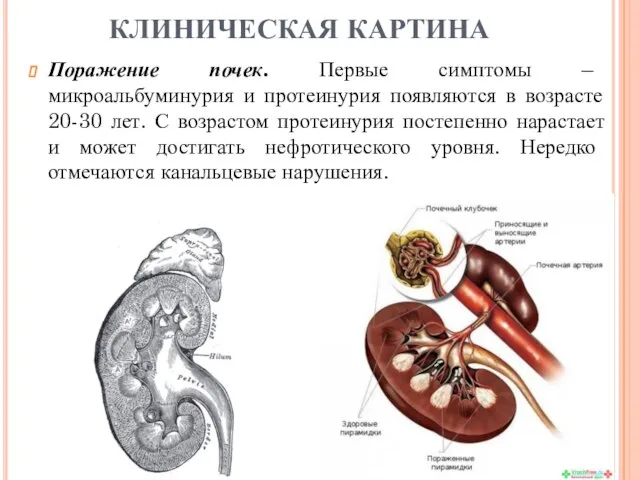
Поражение почек. Первые симптомы – микроальбуминурия и протеинурия появляются в возрасте
Поражение почек. Первые симптомы – микроальбуминурия и протеинурия появляются в возрасте
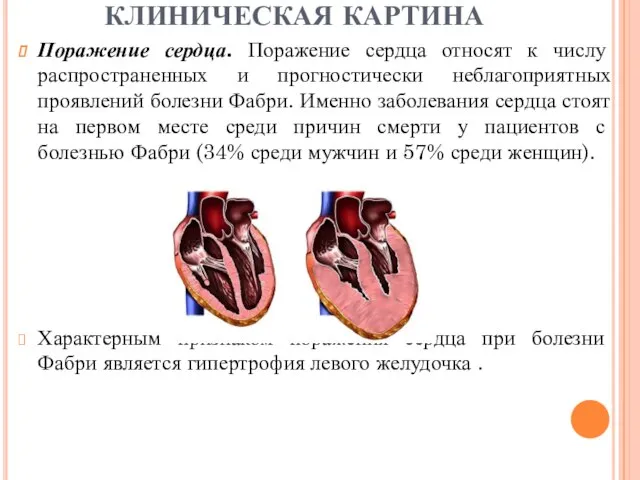
Поражение сердца. Поражение сердца относят к числу распространенных и прогностически неблагоприятных
Поражение сердца. Поражение сердца относят к числу распространенных и прогностически неблагоприятных

Офтальмологические нарушения. Типичным симптомом болезни Фабри является помутнение роговицы в виде
Офтальмологические нарушения. Типичным симптомом болезни Фабри является помутнение роговицы в виде
Изменения со стороны органов слуха и вестибулярного аппарата. У большинства пациентов
Изменения со стороны органов слуха и вестибулярного аппарата. У большинства пациентов

Периферическая нервная система. Болезнь Фабри относится к болезненным полиневропатиям. У пациентов
Периферическая нервная система. Болезнь Фабри относится к болезненным полиневропатиям. У пациентов
Для болезни Фабри типичны болевые кризы, которые часто возникают при перемене
Для болезни Фабри типичны болевые кризы, которые часто возникают при перемене
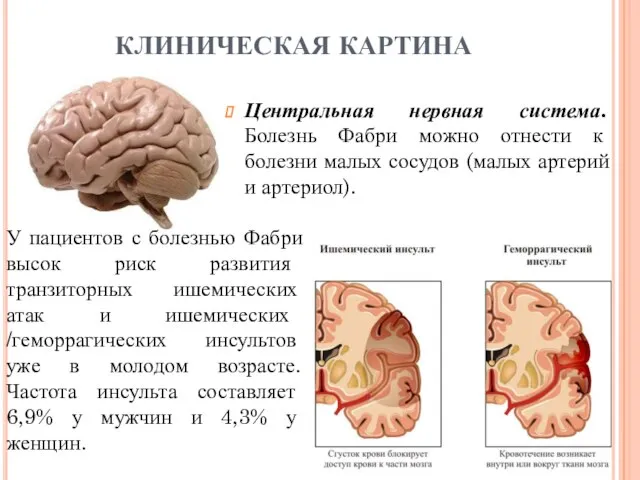
Центральная нервная система. Болезнь Фабри можно отнести к болезни малых сосудов
Центральная нервная система. Болезнь Фабри можно отнести к болезни малых сосудов

У большинства больных инсульт развивается в возрасте от 20 до 50
У большинства больных инсульт развивается в возрасте от 20 до 50

ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Заподозрить болезнь Фабри проще при наличии классического варианта –
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Заподозрить болезнь Фабри проще при наличии классического варианта –

ДНК - ДИАГНОСТИКА
случае неоднозначных результатов биохимических исследований у больных, а также
ДНК - ДИАГНОСТИКА
случае неоднозначных результатов биохимических исследований у больных, а также

ДИАГНОСТИКА
Диагностика болезни Фабри включает комплексную оценку клинический картины, лабораторные тесты и
ДИАГНОСТИКА
Диагностика болезни Фабри включает комплексную оценку клинический картины, лабораторные тесты и

ЛЕЧЕНИЕ ИСПОЛЬЗОВАНИЕ ФЕРМЕНТОЗАМЕСТИТЕЛЬНОЙ ТЕРАПИИ (ФЗТ).
Заместительная терапия рекомбинантными препаратами альфа-галактозидазы А
ЛЕЧЕНИЕ ИСПОЛЬЗОВАНИЕ ФЕРМЕНТОЗАМЕСТИТЕЛЬНОЙ ТЕРАПИИ (ФЗТ).
Заместительная терапия рекомбинантными препаратами альфа-галактозидазы А

СИМПТОМАТИЧЕСКАЯ ТЕРАПИЯ
У пациентов с хронической невропатической болью и акропарестезиями целесообразно по
СИМПТОМАТИЧЕСКАЯ ТЕРАПИЯ
У пациентов с хронической невропатической болью и акропарестезиями целесообразно по

У пациентов с диспепсией возможно лечение блокаторами H2-гистаминовых рецепторов или ингибиторами
У пациентов с диспепсией возможно лечение блокаторами H2-гистаминовых рецепторов или ингибиторами

ПРОГНОЗ
При своевременном доступе к ФЗТ, надлежащем мониторинге болезни и соблюдении врачебных
ПРОГНОЗ
При своевременном доступе к ФЗТ, надлежащем мониторинге болезни и соблюдении врачебных
 Ревматические болезни. Пороки сердца
Ревматические болезни. Пороки сердца Психологическая коррекция самооценки у дошкольников с задержкой психического развития 6-7 лет
Психологическая коррекция самооценки у дошкольников с задержкой психического развития 6-7 лет ГЧП в сфере здравоохранения РФ
ГЧП в сфере здравоохранения РФ Реабилитация при переломе позвоночника
Реабилитация при переломе позвоночника Профилактика йододефицитных состояний
Профилактика йододефицитных состояний Тенденции и особенности отдельных видов заболеваемости в разных регионах мира, странах
Тенденции и особенности отдельных видов заболеваемости в разных регионах мира, странах Сананың бейэпилептикалық параксизмальдық бұзылыстары
Сананың бейэпилептикалық параксизмальдық бұзылыстары Пошехонская центральная районная больница
Пошехонская центральная районная больница b4e9fcd9-dd28-40cb-821c-2c0b542ac37b (1)
b4e9fcd9-dd28-40cb-821c-2c0b542ac37b (1) Вирус кори
Вирус кори Лекарственные растения содержащие тио- и цианогенные гликозиды
Лекарственные растения содержащие тио- и цианогенные гликозиды Наследование групп крови
Наследование групп крови Этиология и патогенез лихорадки
Этиология и патогенез лихорадки Диагностика и коррекция пищевых зависимостей у подростков на примере булимии
Диагностика и коррекция пищевых зависимостей у подростков на примере булимии Пролапс митрального клапана (ПМК)
Пролапс митрального клапана (ПМК) Антибиотики. Антимиробные препараты
Антибиотики. Антимиробные препараты Жаркент қаласындағы А вирусты гепатитімен ауыратын балалар бойынша талдау
Жаркент қаласындағы А вирусты гепатитімен ауыратын балалар бойынша талдау ЖДП дағы дәнекер тін жүйесі ауруларын дәлелді медицина тұрғысынан диагностикалау және емдеу тәсілдері
ЖДП дағы дәнекер тін жүйесі ауруларын дәлелді медицина тұрғысынан диагностикалау және емдеу тәсілдері Диагностика хронических холециститов, гепатитов, цирроза печени
Диагностика хронических холециститов, гепатитов, цирроза печени Оценка свойств цервикальной слизи. Исследование взаимодействия сперматозоидов с цервикальной слизью
Оценка свойств цервикальной слизи. Исследование взаимодействия сперматозоидов с цервикальной слизью Значение деятельности медицинской сестры с пациентами, страдающими атеросклеротическим поражением различной локализации
Значение деятельности медицинской сестры с пациентами, страдающими атеросклеротическим поражением различной локализации Бронхообструктивті синдром
Бронхообструктивті синдром Патология почек
Патология почек Некомпактный миокард левого желудочка в педиатрической практике
Некомпактный миокард левого желудочка в педиатрической практике Инфекционды аурулар кабинеті
Инфекционды аурулар кабинеті Виды ран и правила оказания первой помощи
Виды ран и правила оказания первой помощи Мышечная система
Мышечная система Что говорить? Структура речевых модулей
Что говорить? Структура речевых модулей