Медико-генетическое консультирование. Дородовая диагностика. Подходы к лечению наследственных болезней
- Медико-генетическое консультирование. Дородовая диагностика. Подходы к лечению наследственных болезней

Содержание
- 2. План лекции: План лекции: История медико-генетического консультирования. Цели, методы и задачи медико-генетического консультирования. Этапы медико-генетического консультирования.
- 3. Обращали ли вы внимание на эту часть мозаики?
- 4. Сэр Фрэнсис Гальтон Может с полным правом считаться одним из «отцов» генетики человека Кузен Ч.Дарвина Занимался
- 5. У них с Ч. Дарвином был общий дед – Эразм Дарвин Эра́зм Да́рвин - английский врач,
- 6. Об истории консультирования Первые генетические консультации начали проводиться в США (штат Нью-Йорк) в 1910 г. Чарльзом
- 7. Давиденков Сергей Николаевич крупнейший отечественный невропатолог и генетик человека. Книга «Наследственные болезни нервной системы» (1932). С.Н.Давиденкова
- 8. Медико-генетическое консультирование это специализированный вид медицинской помощи, направленный на предупреждение появления в семьях больных детей. Проспективное
- 9. Потребность населения в МГК. Частота наследственных заболеваний невелика, встречаемость большинства моногенных болезней 1 на сотни тысяч.
- 10. Структура наследственной патологии Три основные группы наследственных заболеваний
- 11. Показаниями для медико-генетического консультирования являются: 1) рождение ребенка с пороком развития; 2) наследственная болезнь в семье;
- 12. В ходе консультации семья должна получить ответы на такие вопросы: Какова природа заболевания? (Не всякое врожденное
- 13. Задачами врача-генетика является: Поставить диагноз Рассчитать генетический риск Донести информацию до семьи Показателем того, что консультация
- 14. Методы, которые используются при консультировании Генеалогический Цитогенетический Биохимический ДНК-диагностики Популяционно-статистический Дерматоглифический
- 15. Этапы медико-генетического консультирования
- 16. Первое посещение врача Сбор генетического анамнеза и построение генеалогического древа Осмотр пробанда (и его родственников) -
- 17. Составление родословной родословную начинают строить с пробанда - лица, с которого начинается исследование семьи. Братья и
- 18. Анализ фенотипа Генетик особое внимание уделяет деталям строения и мелким анатомическим особенностям. У генетиков своя терминология
- 19. Примеры особенностей развития
- 20. Наиболее тщательно изучаются лицо, глаза, Антимонголоидный разрез глаз, гипертелоризм, телекант, гетерохромия радужек Микроцефалия, монголоидный разрез глаз
- 21. строение ротовой полости, микрогнатия макроглоссия олигодонтия Аномальные уздечки во рту «Готическое» нёбо
- 22. строение ушей, Микротия Периаурикулярные выросты Атрезия слухового прохода Периарикулярные ямки Низко посаженные уши Насечки на мочке
- 23. Клинодактилия мизинца Арахнодактилия при синдроме Марфана Камптодактилия Брахидактилия и клинодактилия Брахи- и синдактилия особенности кистей и
- 24. особенности кистей, стоп,
- 25. особенности стоп. Косолапость Сандалевидная щель Стопа-качалка Отечность (при синдроме Шерешевского-Тернера)
- 26. Кожа, ногти, волосы Гемангиома лица при синдроме Штурге-Вебера витилиго Сверхрастяжимость и рубцы типа «папиросной бумаги» при
- 27. Исследуется дерматоглифики Изучаются особенности гребешковой кожи и основные сгибательные линии ладоней и подошв
- 28. Три основных вида пальцевых узоров дуга петля завиток
- 29. Варианты сгибательных складок
- 30. Особенности дерматоглифики при некоторых синдромах Синдром Эдвардса – дуги на всех пальцах Синдром Дауна – одна
- 31. Анализ фенотипа позволяет поставить предварительный диагноз Но иногда требуются дополнительные исследования: Кариотипирование ДНК-диагностика Консультации специалистов (например
- 32. Второе и последующие посещения МГК семьей Постановка окончательного диагноза (в половине случаев не удается поставить диагноз)
- 33. Каковы же основные группы наследственных заболеваний? Моногенные, или менделирующие болезни (см. каталог ОMIM), когда заболевание контролируется
- 34. Структура наследственной патологии Три основные группы наследственных заболеваний
- 35. 1. Моногенные болезни : Ферментопатии – дефекты отдельных ферментов (ФКУ, альбинизм, мукополисахаридозы) Дисплазии – нарушение строения
- 36. Ферментопатии (синдромы дизметаболизма) – дефект отдельного фермента, например: Фенилкетонурия, АР Адреногенитальный синдром, АР Мукополисахаридозы Дети рождаются
- 37. Дисплазии (при мутациях генов, экспрессирующихся в определенных тканях): Нейрофиброматоз (болезнь Реклингаузена), АД * (см. также сл.
- 38. Признаки нейрофиброматоза: фибромы и пятна на коже типа «кофе с молоком». Фибромы происходят из Шванновских клеток.
- 39. Синдромы МВПР http://genetics.rusmedserv.com/syndrom/user/ Множественные врожденные пороки развития - результат мутаций важных регуляторных или генов с плейотропным
- 40. Синдром Беквита-Видеманна это еще и пример нарушенного импринтинга При синдроме Беквита-Видеманна и отцовский и материнский аллели
- 41. Правильный диагноз позволяет предвидеть развитие осложнений. Рассмотрим синдром Беквита-Видеманна Минимальные диагностические признаки: макроглоссия, грыжа пупочного канатика,
- 42. Расчет риска при моногенных болезнях Если заболевание носит семейный характер – исходя из родословной Если это
- 43. Доля новых мутаций для некоторых моногенных болезней: Синдром Беквита-Видемана > 85% Ахондроплазия – 80% Нейрофиброматоз –
- 44. 2. Хромосомные болезни Диагноз базируется на цитогенетическом исследовании. Хромосомные болезни включают: Геномные мутации – изменение числа
- 45. Генетический риск при хромосомных болезнях Новые мутации исходя из популяционного риска Семейные случаи рассчитывается исходя из
- 46. Классический вариант синдрома Дауна (95% случаев) – полная трисомия 46, ХХ 46, ХУ 47,ХУ,21+ Новая мутация
- 47. Семейные транслокации при синдроме Дауна (2 – 3%) Пара 14 Пара 21 Родитель со сбалансированной транслокацией
- 48. При слиянии гамет только один потомок из шести получит нормальный набор хромосом Не совместимы с жизнью
- 49. Бывает риск даже 100% Например, при транслокации 21-й хромосомы на ее гомолог, риск рождения больного ребенка
- 50. 3. Мультифакторные заболевания Обусловлены как генотипом, так и факторами внешней среды. Это наиболее распространенные болезни: ревматизм,
- 51. Схема развития мультифакторного заболевания ген ген ген ген ген ген ген ген среда болезнь генотип
- 52. Пример: Упрощенная схема развития бронхиальной астмы Гены бронхогенной гиперреактивности Гены воспаления Гены атопии Факторы среды: аллергены
- 53. Много ли генов могут отвечать за мультифакторный признак? Вот что пишут про бронхиальную астму:
- 54. Гены, отвечающие за бронхиальную астму
- 55. Гены, отвечающие за бронхиальную астму
- 56. 1900, а? Гены, отвечающие за бронхиальную астму
- 57. Факторы среды, провоцирующие бронхиальную астму пыльца плесень домашние животные пылевые клещи
- 58. Риск в случае мультифакторных болезней рассчитать сложно Обобщенные данные литературы по мультифакториальным заболеваниям собраны в так
- 59. Риск атопии* у пробанда при достижении им возраста 7 лет *Атопия (atopia - греч. нечто необычное,
- 60. Другие гены регуляторы гомеостаза Риск заболевания родственников больных психическими болезнями (в процентах)
- 61. Вывод: для определения риска при моногенных болезнях учитывают родословную при хромосомной патологии учитывают кариотипы родителей при
- 62. Болезни с нетрадиционным наследованием: Цитоплазматическое (митохондриальное) наследование – риск при передаче от матери Болезни геномного импринтинга
- 63. Генетический риск – вероятность появления заболевания у члена семьи Риск развития заболевания менее 5 % считается
- 64. Заключительный этап консультирования – сообщение результатов семье Диагноз и процент риска сообщается только родителям На беседу
- 65. Цель консультирования– принятие родителями адекватного решения Возможные решения родителей: Рожать Не рожать Усыновить Разорвать брак Родить
- 66. Дородовая (пренатальная) диагностика
- 67. Методы дородовой диагностики Неинвазивные методы: Ультразвуковое исследование (все сроки) ХГЧ, альфа-фетопротеин и эстриол в крови матери
- 68. Преимплантационная диагностика При экстракорпоральном оплодотворении (ЭКО) берутся бластомеры на стадии морулы и изучаются до имплантации зародыша.
- 69. Неинвазивные методы УЗИ Исследование сыворотки матери (гормоны и ДНК плода)
- 70. Инвазивные методы
- 71. Биопсия хориона на 8 – 10 неделе беременности
- 72. Кордоцентез – взятие крови из пупочной вены Амниоцентез – взятие околоплодных вод Плацентоцентез – биопсия ткани
- 73. Процедуры проводят под контролем УЗИ
- 74. Каков риск прерывания беременности при инвазивных исследованиях?
- 75. Риск прерывания беременности при дородовой диагностике
- 76. Материал биопсии, полученный при инвазивном заборе исследуют цитогенетически, биохимически, методами ДНК-диагностики. Врач сообщает семье результаты. По
- 77. Диагностика хромосомных болезней FISH-методом
- 78. ДНК-диагностика наследственных болезней - наиболее адекватная и точная диагностика В OMIM описано около 5 тысяч фенотипов
- 79. Лечение наследственных болезней. Возможно?
- 80. Можно выделить 2 подхода к лечению наследственных болезней 1. генная терапия это радикальное устранения генетического дефекта
- 81. Схема генной терапии тяжелого комбинированного иммунодефицита (SCID), вызванного дефектом гена аденозиндезаминазы (АДА) Пример генной терапии Ашани
- 82. Ученые впервые вырастили полноценную кожу и пересадили ее ребенку Семилетний ребенок страдал крайне редким генетическим заболеванием
- 83. Пограничный буллезный эпидермолиз – заболевание, которым в мире болеют около 500 000 человек. Около 40% умирает
- 84. a, Clinical picture of the patient showing massive epidermal loss. b, Schematic representation of the clinical
- 85. Другие фото из статьи
- 87. "Начиная с 7 лет, мы обучаем детей самостоятельно вводить препарат, и, начиная с 10 лет, они
- 88. Диета позволяет избежать проявления признаков фенилкетонурии
- 89. ФКУ диагностируется в роддоме при помощи неонатального скрининга – «просеивания» всех младенцев на наличие биохимических нарушений
- 91. Скачать презентацию

План лекции:
План лекции:
История медико-генетического консультирования.
Цели, методы и задачи медико-генетического консультирования.
Этапы медико-генетического
План лекции:
План лекции:
История медико-генетического консультирования.
Цели, методы и задачи медико-генетического консультирования.
Этапы медико-генетического

Обращали ли вы внимание на эту часть мозаики?
Обращали ли вы внимание на эту часть мозаики?

Сэр Фрэнсис Гальтон
Может с полным правом считаться одним из «отцов» генетики
Сэр Фрэнсис Гальтон
Может с полным правом считаться одним из «отцов» генетики
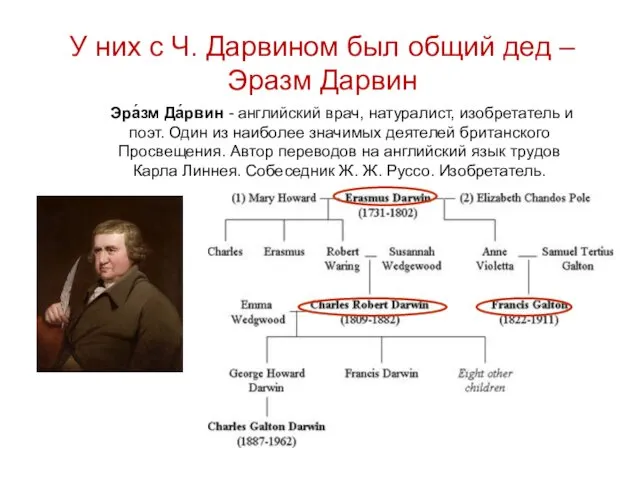
У них с Ч. Дарвином был общий дед – Эразм Дарвин
У них с Ч. Дарвином был общий дед – Эразм Дарвин

Об истории консультирования
Первые генетические консультации начали проводиться в США (штат Нью-Йорк)
Об истории консультирования
Первые генетические консультации начали проводиться в США (штат Нью-Йорк)

Давиденков Сергей Николаевич
крупнейший отечественный невропатолог и генетик человека. Книга «Наследственные болезни
Давиденков Сергей Николаевич
крупнейший отечественный невропатолог и генетик человека. Книга «Наследственные болезни

Медико-генетическое консультирование это
специализированный вид медицинской помощи, направленный на предупреждение появления в
Медико-генетическое консультирование это
специализированный вид медицинской помощи, направленный на предупреждение появления в

Потребность населения в МГК.
Частота наследственных заболеваний невелика, встречаемость большинства моногенных болезней
Потребность населения в МГК.
Частота наследственных заболеваний невелика, встречаемость большинства моногенных болезней
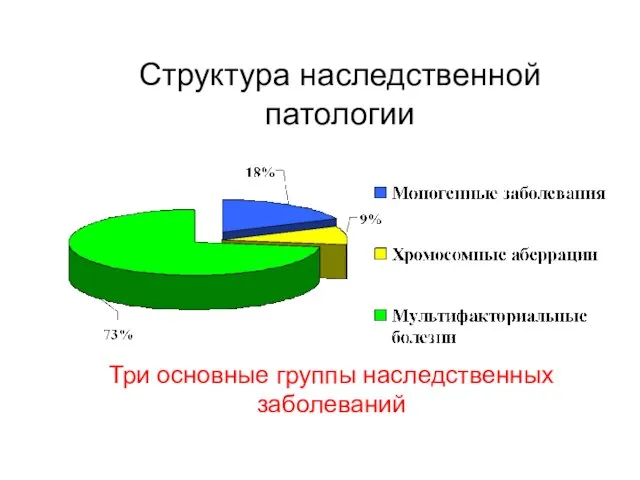
Структура наследственной патологии
Три основные группы наследственных заболеваний
Структура наследственной патологии
Три основные группы наследственных заболеваний

Показаниями для медико-генетического консультирования являются:
1) рождение ребенка с пороком развития;
Показаниями для медико-генетического консультирования являются:
1) рождение ребенка с пороком развития;

В ходе консультации семья должна получить ответы на такие вопросы:
Какова природа
В ходе консультации семья должна получить ответы на такие вопросы:
Какова природа

Задачами врача-генетика является:
Поставить диагноз
Рассчитать генетический риск
Донести информацию до семьи
Показателем
Задачами врача-генетика является:
Поставить диагноз
Рассчитать генетический риск
Донести информацию до семьи
Показателем

Методы, которые используются при консультировании
Генеалогический
Цитогенетический
Биохимический
ДНК-диагностики
Популяционно-статистический
Дерматоглифический
Методы, которые используются при консультировании
Генеалогический
Цитогенетический
Биохимический
ДНК-диагностики
Популяционно-статистический
Дерматоглифический

Этапы медико-генетического консультирования
Этапы медико-генетического консультирования

Первое посещение врача
Сбор генетического анамнеза и построение генеалогического древа
Осмотр пробанда (и
Первое посещение врача
Сбор генетического анамнеза и построение генеалогического древа
Осмотр пробанда (и
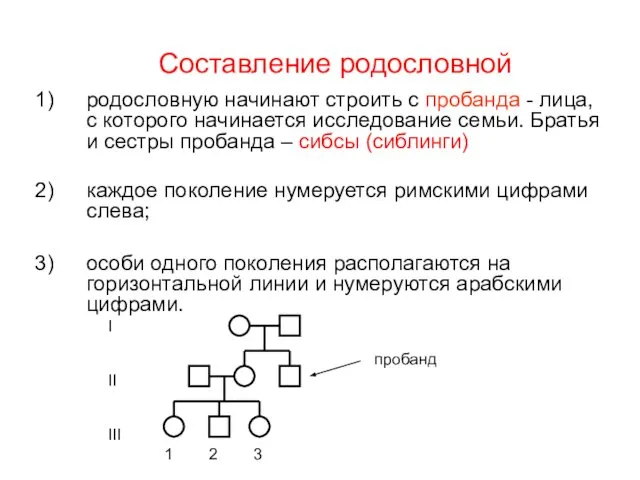
Составление родословной
родословную начинают строить с пробанда - лица, с которого начинается
Составление родословной
родословную начинают строить с пробанда - лица, с которого начинается

Анализ фенотипа
Генетик особое внимание уделяет деталям строения и мелким анатомическим особенностям.
У
Анализ фенотипа
Генетик особое внимание уделяет деталям строения и мелким анатомическим особенностям.
У

Примеры особенностей развития
Примеры особенностей развития
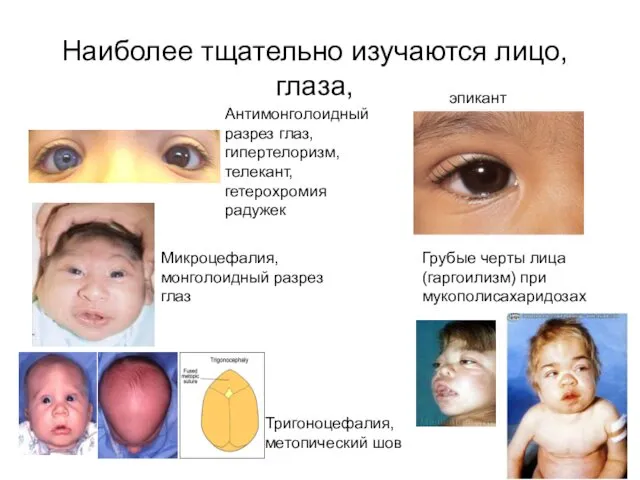
Наиболее тщательно изучаются лицо, глаза,
Антимонголоидный разрез глаз, гипертелоризм, телекант, гетерохромия радужек
Микроцефалия,
Наиболее тщательно изучаются лицо, глаза,
Антимонголоидный разрез глаз, гипертелоризм, телекант, гетерохромия радужек
Микроцефалия,
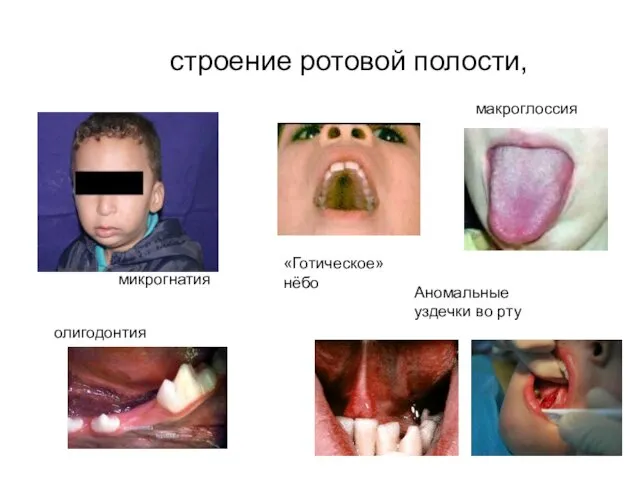
строение ротовой полости,
микрогнатия
макроглоссия
олигодонтия
Аномальные уздечки во рту
«Готическое» нёбо
строение ротовой полости,
микрогнатия
макроглоссия
олигодонтия
Аномальные уздечки во рту
«Готическое» нёбо
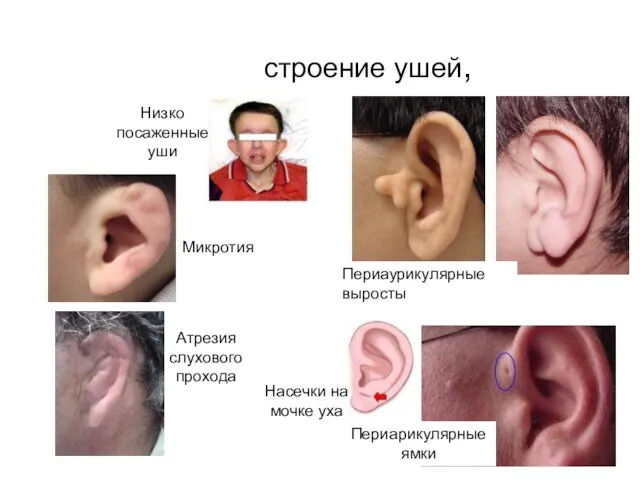
строение ушей,
Микротия
Периаурикулярные выросты
Атрезия слухового прохода
Периарикулярные ямки
Низко посаженные уши
Насечки на мочке
строение ушей,
Микротия
Периаурикулярные выросты
Атрезия слухового прохода
Периарикулярные ямки
Низко посаженные уши
Насечки на мочке
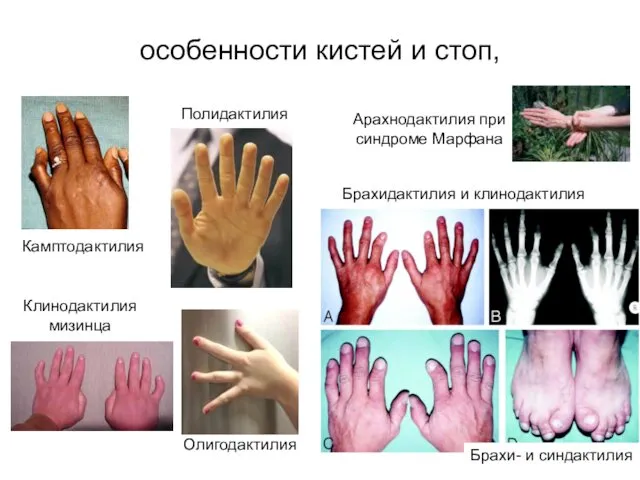
Клинодактилия мизинца
Арахнодактилия при синдроме Марфана
Камптодактилия
Брахидактилия и клинодактилия
Брахи- и синдактилия
особенности
Клинодактилия мизинца
Арахнодактилия при синдроме Марфана
Камптодактилия
Брахидактилия и клинодактилия
Брахи- и синдактилия
особенности
особенности кистей, стоп,
особенности кистей, стоп,
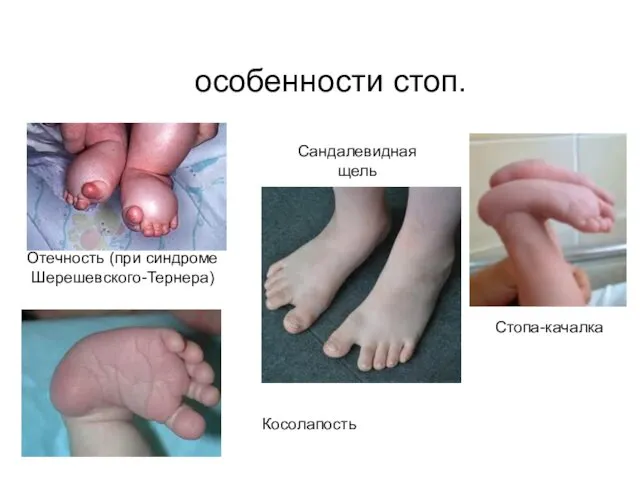
особенности стоп.
Косолапость
Сандалевидная щель
Стопа-качалка
Отечность (при синдроме Шерешевского-Тернера)
особенности стоп.
Косолапость
Сандалевидная щель
Стопа-качалка
Отечность (при синдроме Шерешевского-Тернера)
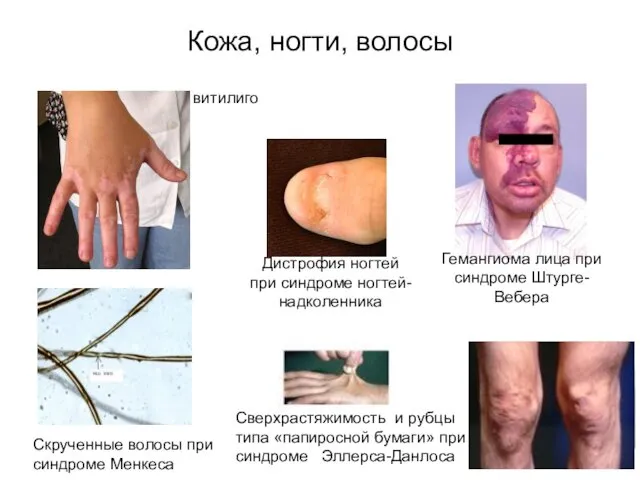
Кожа, ногти, волосы
Гемангиома лица при синдроме Штурге-Вебера
витилиго
Сверхрастяжимость и рубцы типа «папиросной
Кожа, ногти, волосы
Гемангиома лица при синдроме Штурге-Вебера
витилиго
Сверхрастяжимость и рубцы типа «папиросной

Исследуется дерматоглифики
Изучаются особенности гребешковой кожи и основные сгибательные линии ладоней и
Исследуется дерматоглифики
Изучаются особенности гребешковой кожи и основные сгибательные линии ладоней и
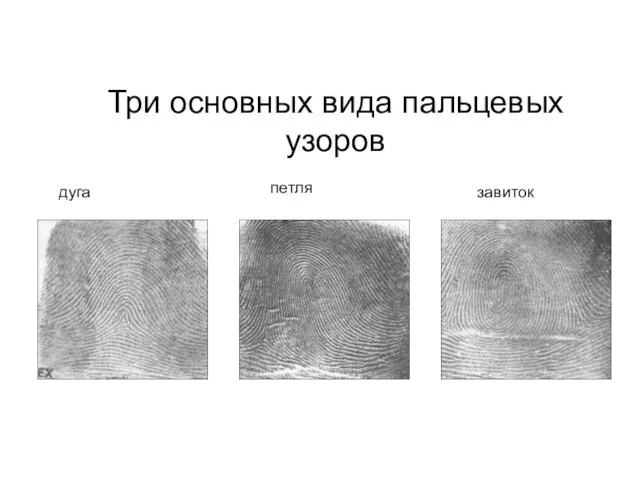
Три основных вида пальцевых узоров
дуга
петля
завиток
Три основных вида пальцевых узоров
дуга
петля
завиток
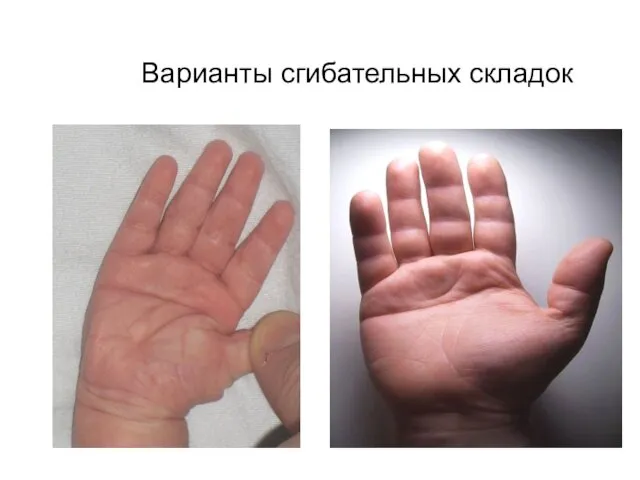
Варианты сгибательных складок
Варианты сгибательных складок

Особенности дерматоглифики при некоторых синдромах
Синдром Эдвардса – дуги на всех пальцах
Синдром
Особенности дерматоглифики при некоторых синдромах
Синдром Эдвардса – дуги на всех пальцах
Синдром

Анализ фенотипа позволяет поставить предварительный диагноз
Но иногда требуются дополнительные исследования:
Кариотипирование
ДНК-диагностика
Консультации специалистов
Анализ фенотипа позволяет поставить предварительный диагноз
Но иногда требуются дополнительные исследования:
Кариотипирование
ДНК-диагностика
Консультации специалистов

Второе и последующие посещения МГК семьей
Постановка окончательного диагноза (в половине случаев
Второе и последующие посещения МГК семьей
Постановка окончательного диагноза (в половине случаев

Каковы же основные группы наследственных заболеваний?
Моногенные, или менделирующие болезни (см. каталог
Каковы же основные группы наследственных заболеваний?
Моногенные, или менделирующие болезни (см. каталог
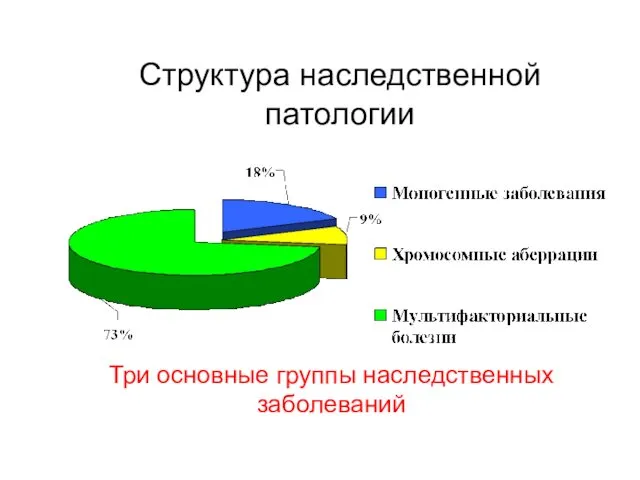
Структура наследственной патологии
Три основные группы наследственных заболеваний
Структура наследственной патологии
Три основные группы наследственных заболеваний

1. Моногенные болезни :
Ферментопатии – дефекты отдельных ферментов (ФКУ, альбинизм, мукополисахаридозы)
Дисплазии
1. Моногенные болезни :
Ферментопатии – дефекты отдельных ферментов (ФКУ, альбинизм, мукополисахаридозы)
Дисплазии

Ферментопатии
(синдромы дизметаболизма)
– дефект отдельного фермента, например:
Фенилкетонурия, АР
Адреногенитальный синдром, АР
Мукополисахаридозы
Дети
Ферментопатии
(синдромы дизметаболизма)
– дефект отдельного фермента, например:
Фенилкетонурия, АР
Адреногенитальный синдром, АР
Мукополисахаридозы
Дети
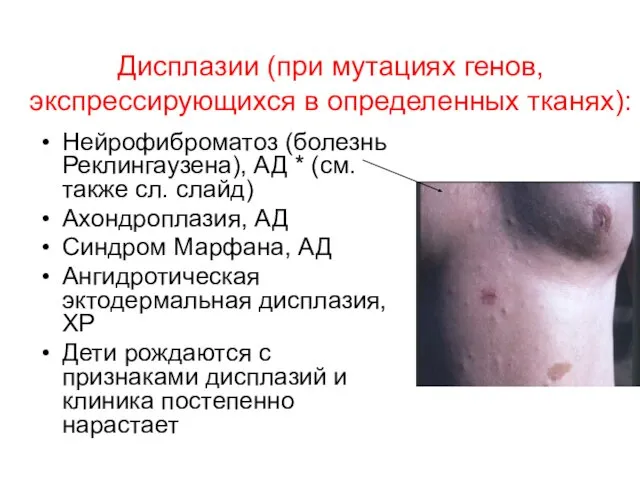
Дисплазии (при мутациях генов, экспрессирующихся в определенных тканях):
Нейрофиброматоз (болезнь Реклингаузена), АД
Дисплазии (при мутациях генов, экспрессирующихся в определенных тканях):
Нейрофиброматоз (болезнь Реклингаузена), АД
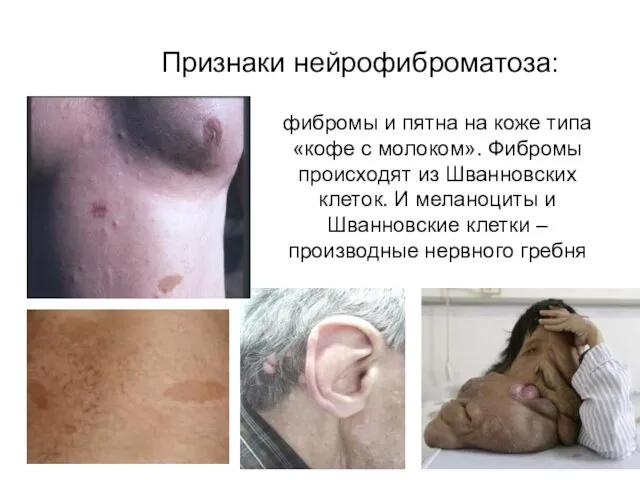
Признаки нейрофиброматоза:
фибромы и пятна на коже типа «кофе с молоком».
Признаки нейрофиброматоза:
фибромы и пятна на коже типа «кофе с молоком».

Синдромы МВПР
http://genetics.rusmedserv.com/syndrom/user/
Множественные врожденные пороки развития - результат мутаций важных регуляторных
Синдромы МВПР
http://genetics.rusmedserv.com/syndrom/user/
Множественные врожденные пороки развития - результат мутаций важных регуляторных
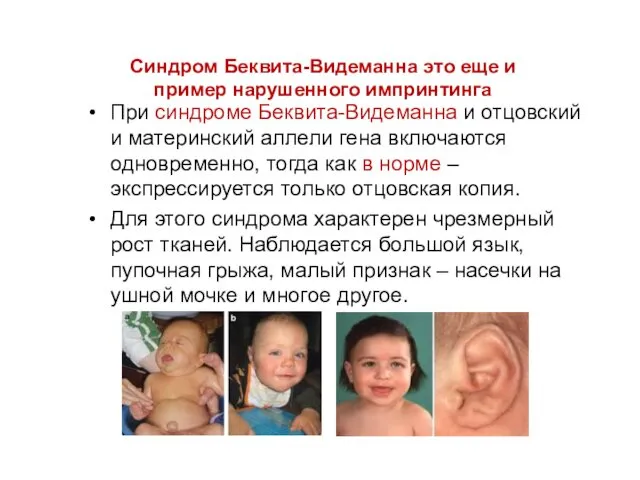
Синдром Беквита-Видеманна это еще и пример нарушенного импринтинга
При синдроме Беквита-Видеманна и
Синдром Беквита-Видеманна это еще и пример нарушенного импринтинга
При синдроме Беквита-Видеманна и

Правильный диагноз позволяет предвидеть развитие осложнений.
Рассмотрим синдром Беквита-Видеманна
Минимальные диагностические признаки:
Правильный диагноз позволяет предвидеть развитие осложнений.
Рассмотрим синдром Беквита-Видеманна
Минимальные диагностические признаки:
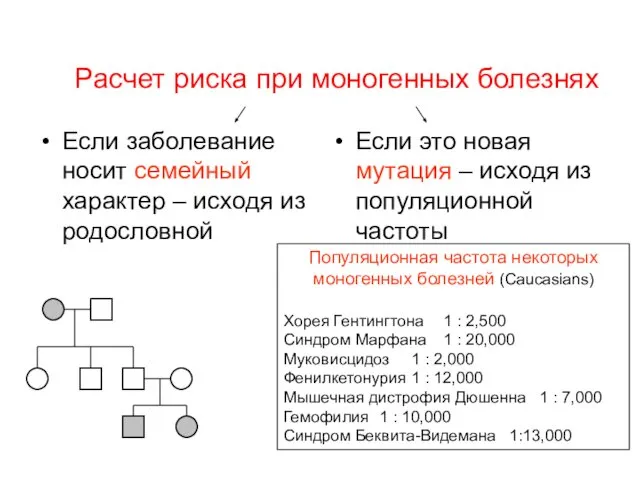
Расчет риска при моногенных болезнях
Если заболевание носит семейный характер – исходя
Расчет риска при моногенных болезнях
Если заболевание носит семейный характер – исходя

Доля новых мутаций для некоторых моногенных болезней:
Синдром Беквита-Видемана > 85%
Ахондроплазия –
Доля новых мутаций для некоторых моногенных болезней:
Синдром Беквита-Видемана > 85%
Ахондроплазия –

2. Хромосомные болезни
Диагноз базируется на цитогенетическом исследовании.
Хромосомные болезни включают:
Геномные мутации –
2. Хромосомные болезни
Диагноз базируется на цитогенетическом исследовании.
Хромосомные болезни включают:
Геномные мутации –

Генетический риск при хромосомных болезнях
Новые мутации исходя из популяционного риска
Семейные случаи
Генетический риск при хромосомных болезнях
Новые мутации исходя из популяционного риска
Семейные случаи
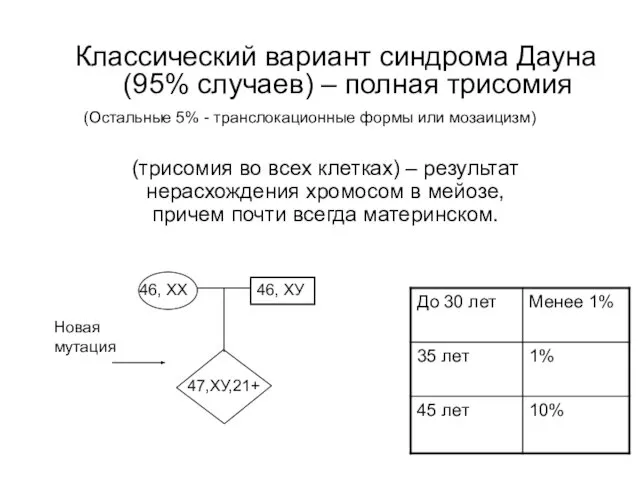
Классический вариант синдрома Дауна (95% случаев) – полная трисомия
46, ХХ
46, ХУ
47,ХУ,21+
Новая
Классический вариант синдрома Дауна (95% случаев) – полная трисомия
46, ХХ
46, ХУ
47,ХУ,21+
Новая
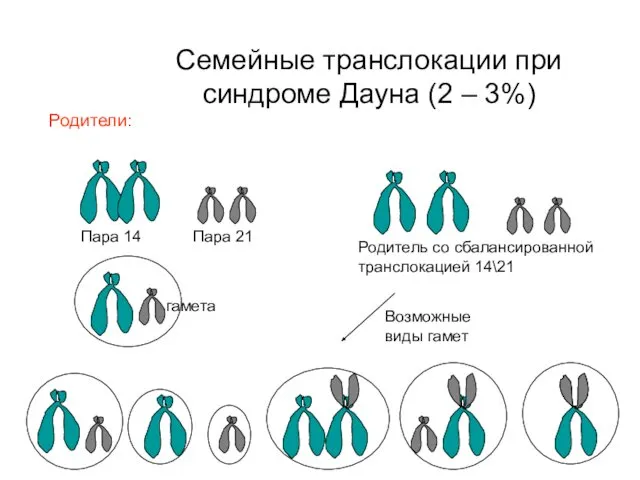
Семейные транслокации при синдроме Дауна (2 – 3%)
Пара 14
Пара 21
Родитель со
Семейные транслокации при синдроме Дауна (2 – 3%)
Пара 14
Пара 21
Родитель со
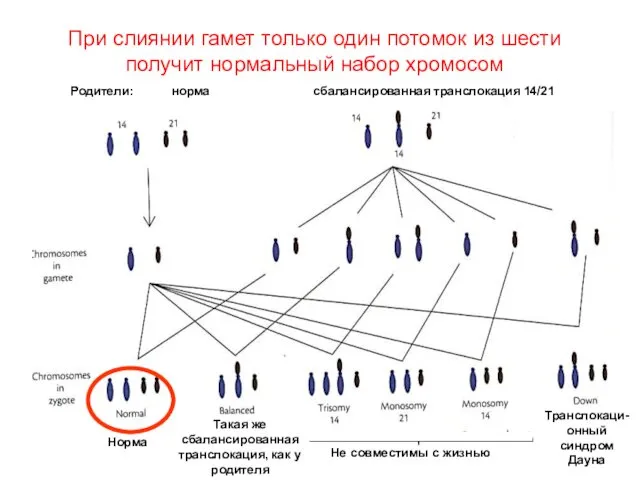
При слиянии гамет только один потомок из шести получит нормальный набор
При слиянии гамет только один потомок из шести получит нормальный набор
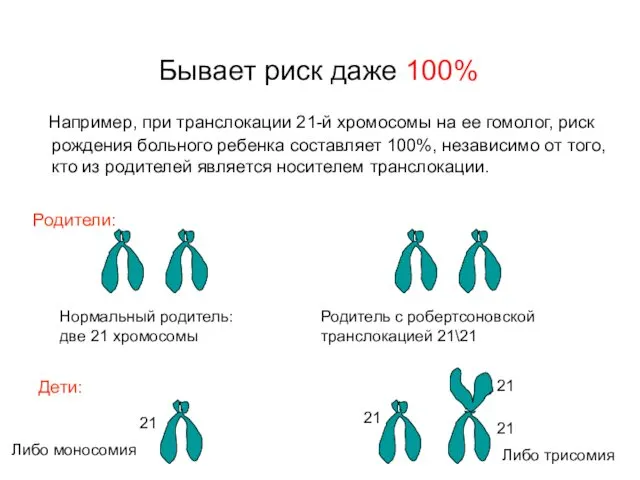
Бывает риск даже 100%
Например, при транслокации 21-й хромосомы на ее
Бывает риск даже 100%
Например, при транслокации 21-й хромосомы на ее

3. Мультифакторные заболевания
Обусловлены как генотипом, так и факторами внешней среды.
Это
3. Мультифакторные заболевания
Обусловлены как генотипом, так и факторами внешней среды.
Это
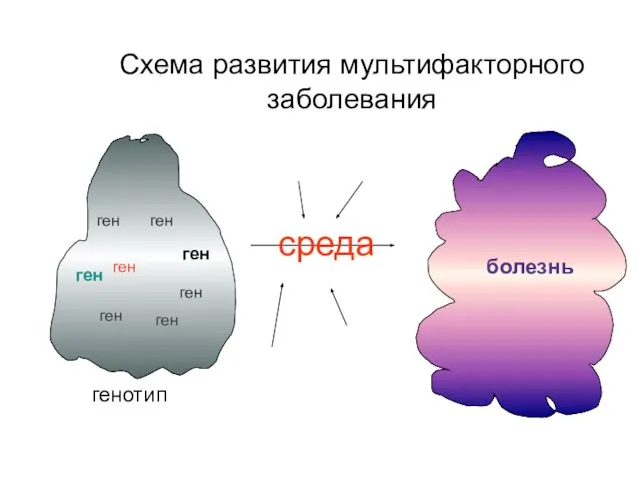
Схема развития мультифакторного заболевания
ген
ген
ген
ген
ген
ген
ген
ген
среда
болезнь
генотип
Схема развития мультифакторного заболевания
ген
ген
ген
ген
ген
ген
ген
ген
среда
болезнь
генотип
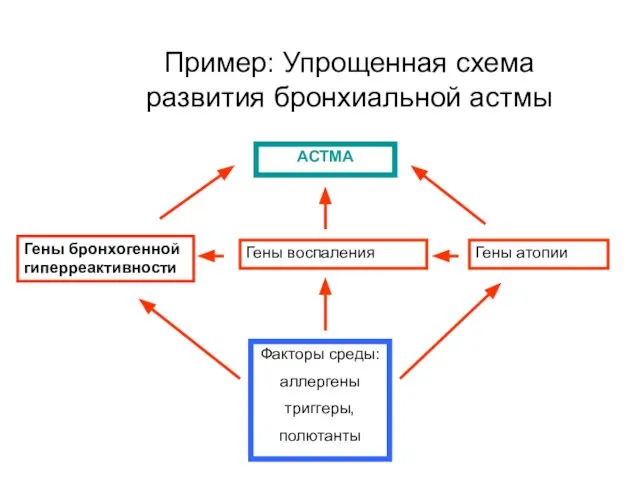
Пример: Упрощенная схема развития бронхиальной астмы
Гены бронхогенной гиперреактивности
Гены воспаления
Гены атопии
Факторы среды:
аллергены
триггеры,
Пример: Упрощенная схема развития бронхиальной астмы
Гены бронхогенной гиперреактивности
Гены воспаления
Гены атопии
Факторы среды:
аллергены
триггеры,

Много ли генов могут отвечать за мультифакторный признак?
Вот что пишут про
Много ли генов могут отвечать за мультифакторный признак? Вот что пишут про
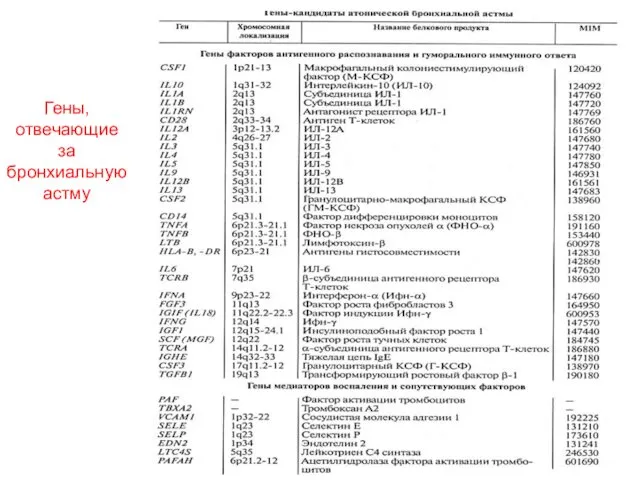
Гены, отвечающие за бронхиальную астму
Гены, отвечающие за бронхиальную астму
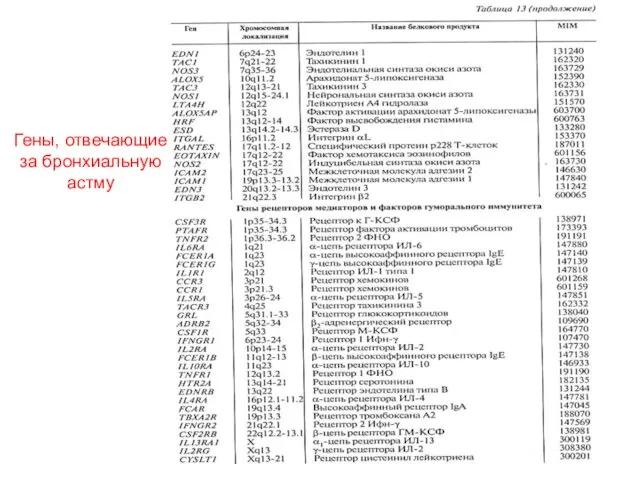
Гены, отвечающие за бронхиальную астму
Гены, отвечающие за бронхиальную астму
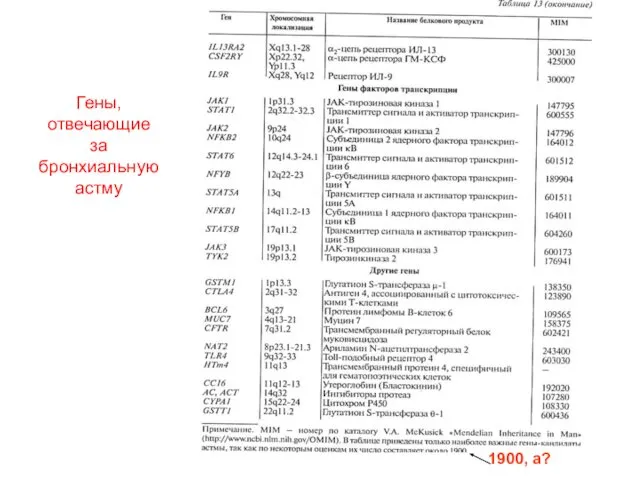
1900, а?
Гены, отвечающие за бронхиальную астму
1900, а?
Гены, отвечающие за бронхиальную астму
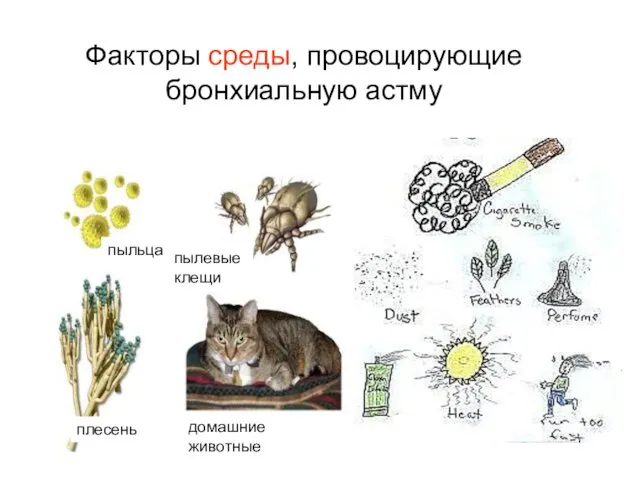
Факторы среды, провоцирующие бронхиальную астму
пыльца
плесень
домашние животные
пылевые клещи
Факторы среды, провоцирующие бронхиальную астму
пыльца
плесень
домашние животные
пылевые клещи

Риск в случае мультифакторных болезней рассчитать сложно
Обобщенные данные литературы по мультифакториальным
Риск в случае мультифакторных болезней рассчитать сложно
Обобщенные данные литературы по мультифакториальным
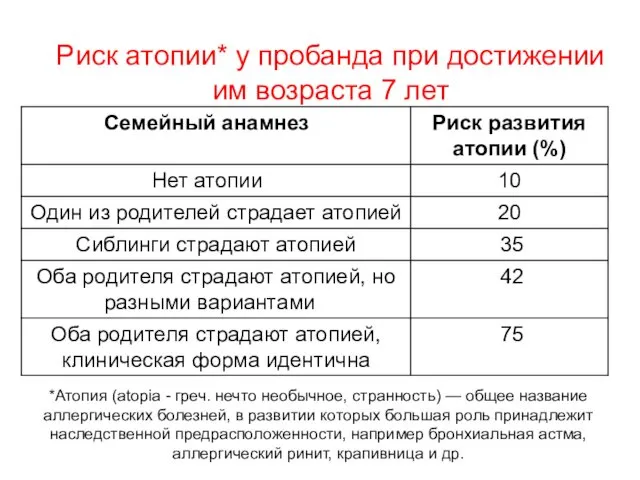
Риск атопии* у пробанда при достижении им возраста 7 лет
*Атопия (atopia
Риск атопии* у пробанда при достижении им возраста 7 лет
*Атопия (atopia
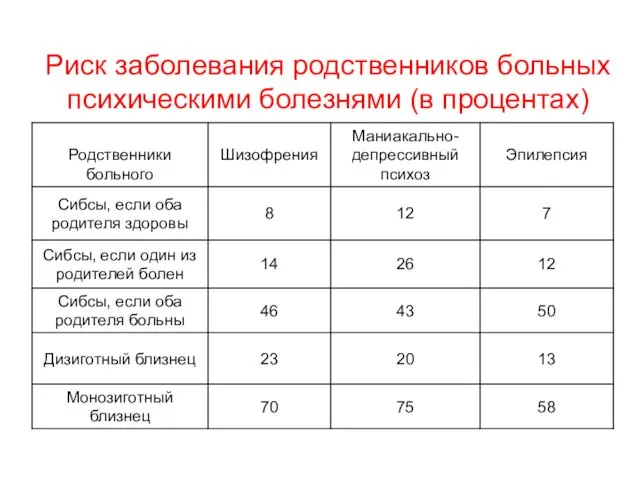
Другие гены регуляторы гомеостаза
Риск заболевания родственников больных психическими болезнями (в процентах)
Другие гены регуляторы гомеостаза
Риск заболевания родственников больных психическими болезнями (в процентах)

Вывод:
для определения риска
при моногенных болезнях учитывают родословную
при хромосомной патологии учитывают
Вывод:
для определения риска
при моногенных болезнях учитывают родословную
при хромосомной патологии учитывают
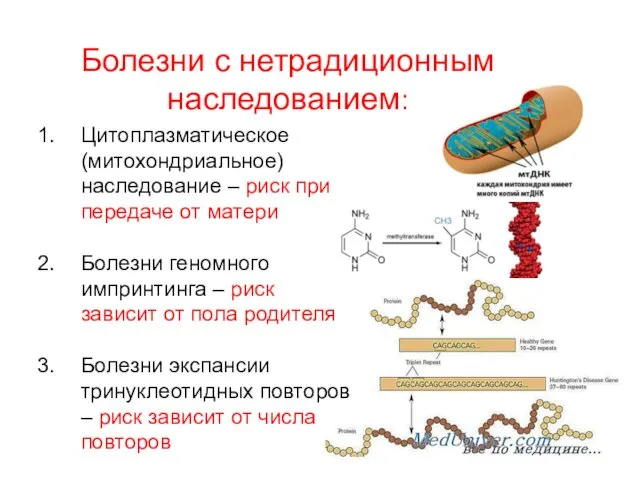
Болезни с нетрадиционным наследованием:
Цитоплазматическое (митохондриальное) наследование – риск при передаче от
Болезни с нетрадиционным наследованием:
Цитоплазматическое (митохондриальное) наследование – риск при передаче от

Генетический риск – вероятность появления заболевания у члена семьи
Риск развития
Генетический риск – вероятность появления заболевания у члена семьи
Риск развития

Заключительный этап консультирования – сообщение результатов семье
Диагноз и процент риска сообщается
Заключительный этап консультирования – сообщение результатов семье
Диагноз и процент риска сообщается

Цель консультирования– принятие родителями адекватного решения
Возможные решения родителей:
Рожать
Не рожать
Усыновить
Разорвать
Цель консультирования– принятие родителями адекватного решения
Возможные решения родителей:
Рожать
Не рожать
Усыновить
Разорвать

Дородовая (пренатальная) диагностика
Дородовая (пренатальная) диагностика
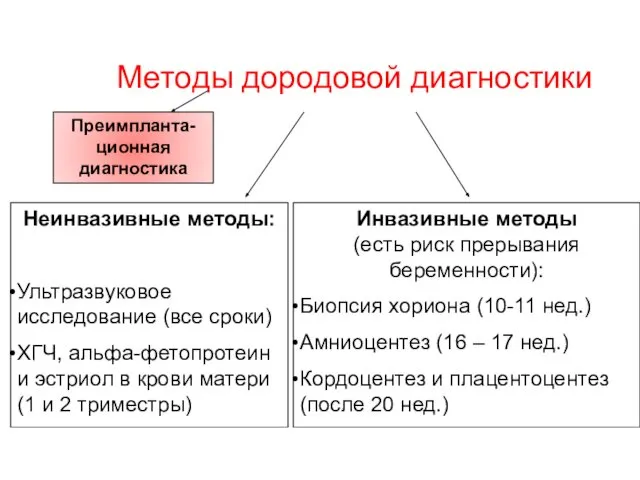
Методы дородовой диагностики
Неинвазивные методы:
Ультразвуковое исследование (все сроки)
ХГЧ, альфа-фетопротеин и эстриол
Методы дородовой диагностики
Неинвазивные методы:
Ультразвуковое исследование (все сроки)
ХГЧ, альфа-фетопротеин и эстриол
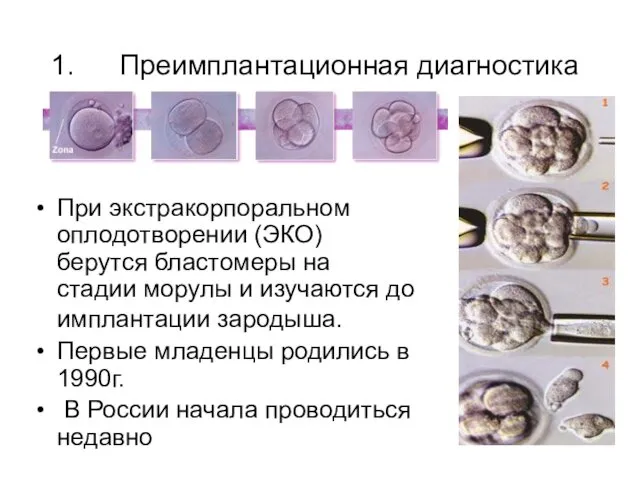
Преимплантационная диагностика
При экстракорпоральном оплодотворении (ЭКО) берутся бластомеры на стадии морулы и
Преимплантационная диагностика
При экстракорпоральном оплодотворении (ЭКО) берутся бластомеры на стадии морулы и

Неинвазивные методы
УЗИ
Исследование сыворотки матери (гормоны и ДНК плода)
Неинвазивные методы
УЗИ
Исследование сыворотки матери (гормоны и ДНК плода)
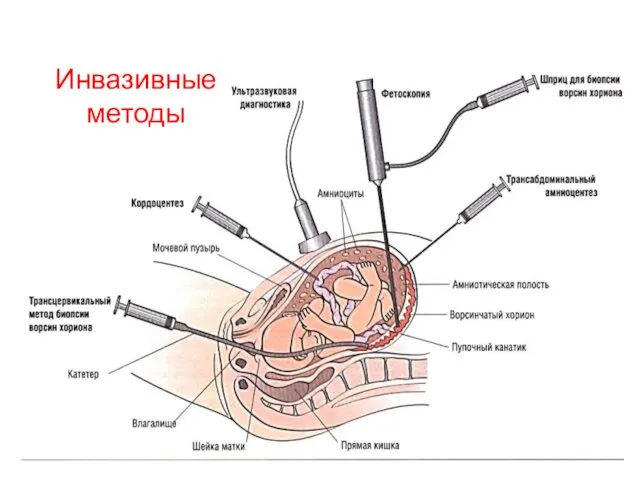
Инвазивные методы
Инвазивные методы
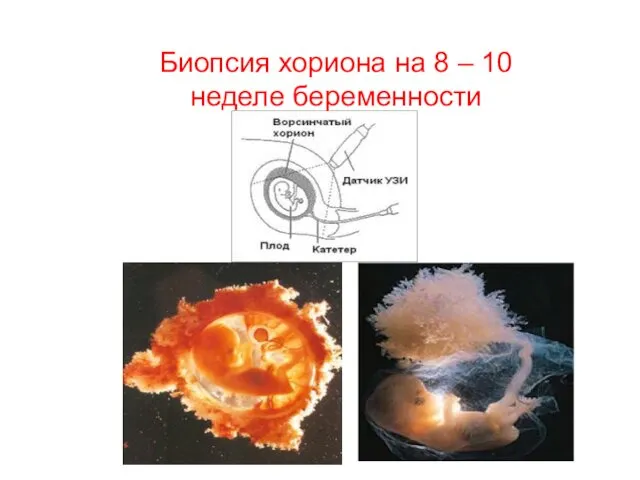
Биопсия хориона на 8 – 10 неделе беременности
Биопсия хориона на 8 – 10 неделе беременности
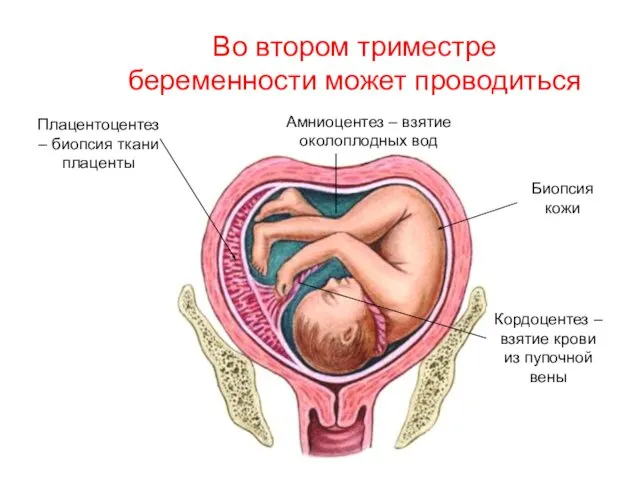
Кордоцентез – взятие крови из пупочной вены
Амниоцентез – взятие околоплодных вод
Плацентоцентез
Кордоцентез – взятие крови из пупочной вены
Амниоцентез – взятие околоплодных вод
Плацентоцентез
Процедуры проводят под контролем УЗИ
Процедуры проводят под контролем УЗИ
Каков риск прерывания беременности при инвазивных исследованиях?
Каков риск прерывания беременности при инвазивных исследованиях?

Риск прерывания беременности при дородовой диагностике
Риск прерывания беременности при дородовой диагностике

Материал биопсии, полученный при инвазивном заборе исследуют
цитогенетически, биохимически, методами ДНК-диагностики.
Врач сообщает
Материал биопсии, полученный при инвазивном заборе исследуют
цитогенетически, биохимически, методами ДНК-диагностики.
Врач сообщает
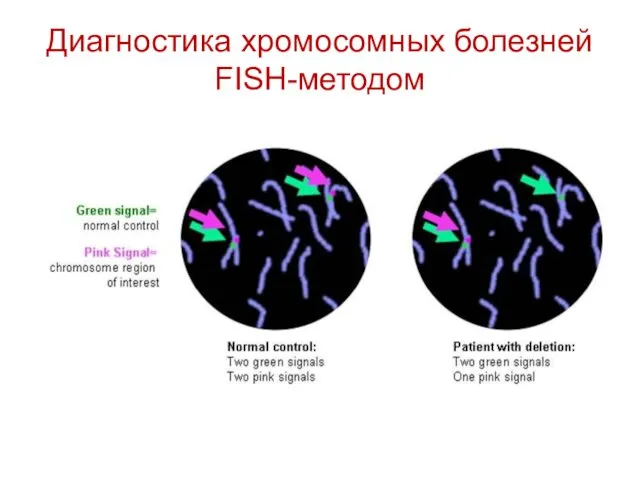
Диагностика хромосомных болезней FISH-методом
Диагностика хромосомных болезней FISH-методом

ДНК-диагностика наследственных болезней - наиболее адекватная и точная диагностика
В OMIM описано
ДНК-диагностика наследственных болезней - наиболее адекватная и точная диагностика
В OMIM описано

Лечение наследственных болезней.
Возможно?
Лечение наследственных болезней.
Возможно?

Можно выделить 2 подхода к лечению наследственных болезней
1. генная терапия это
Можно выделить 2 подхода к лечению наследственных болезней
1. генная терапия это
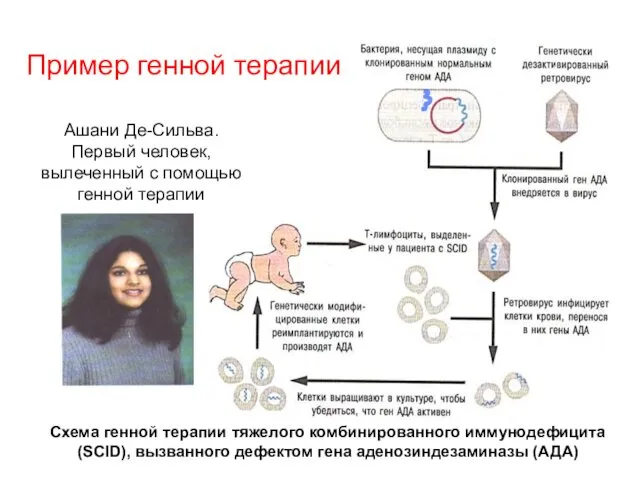
Схема генной терапии тяжелого комбинированного иммунодефицита (SCID), вызванного дефектом гена аденозиндезаминазы
Схема генной терапии тяжелого комбинированного иммунодефицита (SCID), вызванного дефектом гена аденозиндезаминазы

Ученые впервые вырастили полноценную кожу и пересадили ее ребенку
Семилетний ребенок страдал
Ученые впервые вырастили полноценную кожу и пересадили ее ребенку
Семилетний ребенок страдал

Пограничный буллезный эпидермолиз – заболевание, которым в мире болеют около 500
Пограничный буллезный эпидермолиз – заболевание, которым в мире болеют около 500
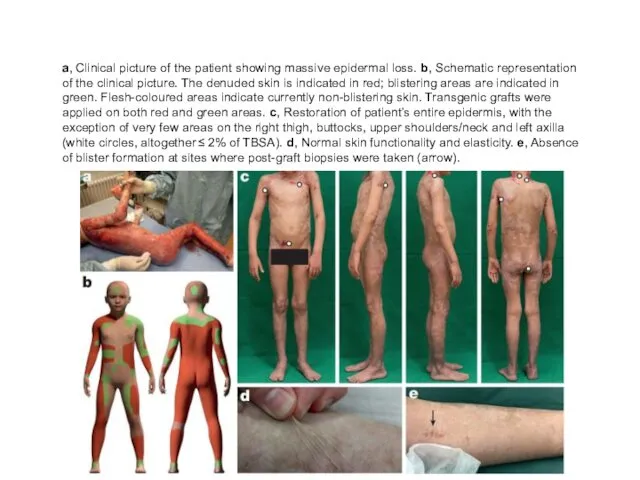
a, Clinical picture of the patient showing massive epidermal loss. b,
a, Clinical picture of the patient showing massive epidermal loss. b,
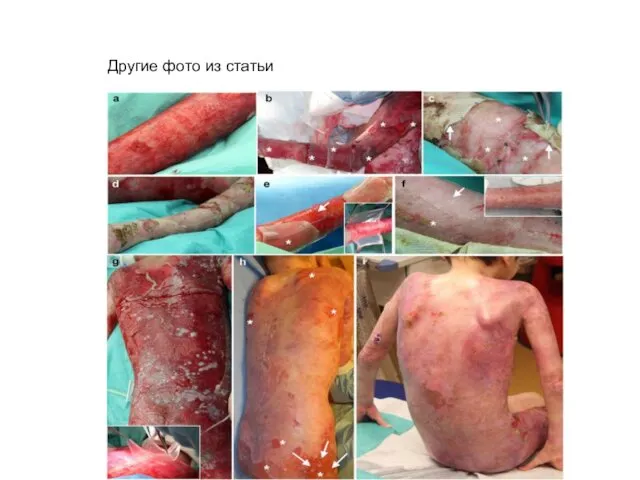
Другие фото из статьи
Другие фото из статьи

"Начиная с 7 лет, мы обучаем детей самостоятельно вводить препарат, и,
"Начиная с 7 лет, мы обучаем детей самостоятельно вводить препарат, и,

Диета позволяет избежать проявления признаков фенилкетонурии
Диета позволяет избежать проявления признаков фенилкетонурии

ФКУ диагностируется в роддоме при помощи неонатального скрининга –
«просеивания» всех
ФКУ диагностируется в роддоме при помощи неонатального скрининга –
«просеивания» всех
 Йога – учение о познании себя и окружающего мира
Йога – учение о познании себя и окружающего мира Антиген
Антиген Босанғаннан кейінгі ерте кезеңдегі қан кетудің себептері: тонус, тін
Босанғаннан кейінгі ерте кезеңдегі қан кетудің себептері: тонус, тін Развитие в юношеском возрасте
Развитие в юношеском возрасте Введение в специальность «Инфекционные болезни». Лекция №1
Введение в специальность «Инфекционные болезни». Лекция №1 Европейский проект Stent for life. Начало и первые результаты в РБ. Ведение пациентов с ОКС на амбулаторном этапе
Европейский проект Stent for life. Начало и первые результаты в РБ. Ведение пациентов с ОКС на амбулаторном этапе Скарлатина. Характеристика возбудителя
Скарлатина. Характеристика возбудителя Внутривенная инъекция. Взятие крови на исследования. Аутогемотерапия
Внутривенная инъекция. Взятие крови на исследования. Аутогемотерапия Аурухана ішілік инфекцияны бақылауды ұйымдастыру
Аурухана ішілік инфекцияны бақылауды ұйымдастыру Хронический гранулирующий периодонтит. Особенности клинического течения и диагностики. Практическое занятие №5
Хронический гранулирующий периодонтит. Особенности клинического течения и диагностики. Практическое занятие №5 Сонная болезнь
Сонная болезнь Система органов кроветворения
Система органов кроветворения Лечебные травы
Лечебные травы Близорукость и дальнозоркость. Операция по восстановлению зрения
Близорукость и дальнозоркость. Операция по восстановлению зрения Тримедат (тримебутин). Клиническая практика и исследования. Новые подходы к восстановлению моторики ЖКТ
Тримедат (тримебутин). Клиническая практика и исследования. Новые подходы к восстановлению моторики ЖКТ Психологическая уравновешенность. Стресс и его влияние на человека
Психологическая уравновешенность. Стресс и его влияние на человека Желудочно-кишечные кровотечения
Желудочно-кишечные кровотечения Протоночная цитометрия
Протоночная цитометрия Кишечная непроходимость
Кишечная непроходимость Психология цвета «Красный»
Психология цвета «Красный» Развитие памяти у младшего школьного возраста 7-10 лет
Развитие памяти у младшего школьного возраста 7-10 лет Клиническийслучай
Клиническийслучай Холера
Холера Артериальная гипертензия
Артериальная гипертензия Familia retroviridae. Virusul HIV. Oncogeneza virala
Familia retroviridae. Virusul HIV. Oncogeneza virala Геморрагиялық шок. ТІШҚҰ-синдромы
Геморрагиялық шок. ТІШҚҰ-синдромы Оценка результатов лечения детей с тератомами крестцово-копчиковой области
Оценка результатов лечения детей с тератомами крестцово-копчиковой области Медицинская стерилизация
Медицинская стерилизация