- Миелопролиферативные заболевания

Содержание
- 2. Миелопролиферативные заболевания представляют собой клональные заболевания, возникающие на уровне стволовой кроветворной клетки, характеризуются пролиферацией одной или
- 3. Созревание в ККМ Созревание в лимфоидных тканях
- 4. КЛАССИФИКАЦИЯ МИЕЛОИДНЫХ НЕОПЛАЗИЙ (ВСЕМИРНАЯ ОРГАНИЗАЦИЯ ЗДРАВООХРАНЕНИЯ, ПЕРЕСМОТР 2016) 1. Миелопролиферативные заболевания (МПЗ) a. Хронический миелолейкоз (ХМЛ),
- 5. Хронические миелопролиферативные заболевания 1. Хронический миелоидный лейкоз (Ph+)(филадельфийская хромосома) (Ph-): 2. Истинная полицитемия (ИП) 3. Эссенциальная
- 6. Хронический миелоидный лейкоз Злокачественная опухоль кроветворной ткани, исходящая из клеток-предшественниц миелопоэза, морфологическим субстратом которой являются дифференцирующиеся
- 7. ХМЛ является клональным миелопролиферативным заболеванием, развивающимся в результате злокачественной трансформации в ранних гемопоэтических предшественниках. Уникальная особенность
- 8. Классификация хронического миелолейкоза (Athens, 1993)
- 9. Хронический миелолейкоз на начальных стадиях протекает бессимптомно. Спустя некоторое время у больных появляется быстрая утомляемость, потливость,
- 10. Спленомегалия выявляется у 90-95% больных Гепатомегалия наблюдается у 50-60% больных
- 11. По мере накопления лейкемического клона, нарастания гепато- и спленомегалии, подавления нормального кроветворения, появляется неспецифическая клиническая симптоматика,
- 12. Общий анализ крови Лейкоцитоз до 50-300 × 10 /л Сдвиг лейкоцитарной формулы влево с появлением молодых
- 13. Миелограмма Повышено содержание миелокариоцитов, мегакариоцитов, гранулоцитов Определяются все элементы гранулоцитарного ряда Число митозов увеличено в 4-5
- 14. Бластный криз Выраженная анемия Лейкоцитоз Нейтропения Бластемия 30% и более «Провал» в лейкоцитарной формуле Тромбоцитопения Увеличение
- 15. Фазы ХМЛ по классификациям ELN
- 16. Цель современной терапии ХМЛ максимальное подавление Ph-положительного опухолевого клона, снижение риска прогрессии заболевания, достижение продолжительности жизни
- 17. В период обследования, до получения результатов цитогенетического исследования, подтверждающих наличие Ph-хромосомы в клетках костного мозга, больному
- 18. Современные направления терапии ХМЛ: Альфа-интерферон антагонист ростовых факторов, антипролиферативная активность, стимуляция противоопухолевого иммунитета цитогенетические ремиссии у
- 19. После подтверждения диагноза ХМЛ должна быть начата терапия ИТК (ингибиторами BCR-ABL-тирозинкиназы). Лечение может проводиться в амбулаторных
- 20. Аллогенная трансплантация гемопоэтических стволовых клеток (алло-ТГСК) должна быть обязательно рассмотрена для больных ХМЛ ХФ с высокой
- 21. Истинная полицитемия (ИП) син.: эритремия, болезнь Вакеза, истинная красная полицитемия -- клональное МПЗ, которое характеризуется пролиферацией
- 22. Клиническая картина Плеторический синдром Головная боль Головокружение Нарушение зрения Кроличьи глаза Боли в области сердца Боли
- 23. Признаки, типичные для полицитемии: гепатомегалия и спленомегалия, гиперемия кожи, гипертензия, ночная потливость, головная боль, кожный зуд,
- 24. Сосудистые осложнения при эритремии Инфаркт миокарда Инсульт Перемежающаяся хромота Гангрена ТЭЛА Тромбоз мезентериальных сосудов Синдром Бадда-Хиари
- 25. Общий анализ крови Повышение гемоглобина до 180-220 г/л Увеличение количества эритроцитов до 6-15 × 10 /л
- 26. Миелограмма Трехростковая гиперплазия с преобладанием эритроидного и мегакариоцитарного ростков Уменьшение количества жировой ткани Снижение лейко-эритроцитарного индекса
- 27. Лабораторная диагностика Биохимический анализ крови повышение содержания мочевой кислоты Радиологические методы увеличение объема циркулирующих эртроцитов, усиление
- 28. Стадии эритремии I стадия – начальная II стадия – эритремическая II A – без миелоидной метаплазии
- 29. Критерии для диагностики истинной полицитемии Большие критерии Hb более 16,5 г/мл для мужчин, Hb более 16,0
- 30. Вторичные эритроцитозы Абсолютные Вследствие генерализованной тканевой гипоксии высотная болезнь, обструктивные заболевания легких, пороки сердца, первичная легочная
- 31. Лечение эритремии Кровопускание 400-500 мл через день, гирудотерапия Дезагреганты аспирин, тиклид, трентал, реополиглюкин Цитостатическая терапия гидреа,
- 32. Суммированные рекомендации по лечению ИП 1. Для всех больных: • кровопускания/эритроцитаферез для поддержания гематокрита в пределах
- 33. 2. Для больных группы низкого риска развития тромбогеморрагических осложнений. • Циторедуктивная терапия показана в случаях: --
- 34. Эссенциальная тромбоцитемия (ЭТ) (син.: первичный тромбоцитоз, идиопатический тромбоцитоз, геморрагическая тромбоцитемия) – клональное МПЗ с неконтролируемой пролиферацией
- 35. Критерии диагностики эссенциальной тромбоцитемии Большие критерии Количество тромбоцитов 450·109 /л и более. При гистологическом исследовании костного
- 36. Эссенциальная тромбоцитемия проявляется обмороками и предобморочными состояниями, болью в груди, пульсирующей боль в кистях или ступнях,
- 37. Суммированные рекомендации при ЭТ 1 Для всех больных: -- профилактика сердечно-сосудистых заболеваний (устранение факторов риска); --
- 38. Сублейкемический миелоз (идиопатический миелофиброз) Хроническое клональное миелопролиферативное заболевание, развивающееся вследствие нарушений стволовой кроветворной клетки и характеризующееся
- 39. Критерии диагностики первичного миелофиброза
- 40. Признаки миелофиброза: одышка, слабость, бледность кожи, боль в животе, потеря веса, гепатоспленомегалия, беспричинные кровотечения, гипергидроз, лихорадка,
- 41. Клиническая картина общая слабость потливость снижение аппетита похудание боли в костях тяжесть и боли в подреберьях
- 42. Общий анализ крови Нормохромная нормоцитарная анемия Анизоцитоз, пойкилоцитоз, дакриоциты, нормобласты Увеличение ретикулоцитов Умеренный лейкоцитоз Миелоциты и
- 43. Миелограмма I стадия: гиперплазия всех трех ростков кроветворения, особенно мегакариоцитарного, очаги ретикулинового миелофиброза II стадия: уменьшение
- 44. В пересмотренной Классификации ВОЗ (2017) подчеркивается важность дифференциальной диагностики эссенциальной тромбоцитемии и префиброзной/ранней стадии первичного миелофиброза
- 45. Методы терапии ПМФ Алло-ТГСК; Медикаментозная терапия : цитостатики, эритропоэзстимулирующие агенты, глюкокортикостероиды, андрогены, ингибиторы JAK2 Хирургическое лечение
- 46. Осложнения при ПМФ и тактика их лечения Опухолевая интоксикация Спленомегалия Анемия Инфекционные осложнения (Лейкопения,нейтропения) Тромбоцитопения и
- 47. Бластная фаза МПЗ является терминальной стадией, прогноз неблагоприятный -- средняя продолжительность жизни составляет 6 мес. Выживаемость
- 48. Прогноз Хронический миелолейкоз продолжительность жизни в среднем 3-5 лет, у отдельных больных 7-8 лет Эритремия продолжительность
- 49. «Миелопролиферативное заболевание, неклассифицируемое» Нозологическая форма «Миелопролиферативное заболевание, неклассифицируемое» устанавливается при наличии несомненных клинических и лабораторных признаков
- 51. Мастоцитозы В новой редакции ВОЗ (2017) группа мастоцитозов выведена из группы МПЗ в связи с их
- 52. КЛАССИФИКАЦИЯ МИЕЛОИДНЫХ НЕОПЛАЗИЙ (ВСЕМИРНАЯ ОРГАНИЗАЦИЯ ЗДРАВООХРАНЕНИЯ, ПЕРЕСМОТР 2016) 1. Миелопролиферативные заболевания (МПЗ) a. Хронический миелолейкоз (ХМЛ),
- 53. Миелодиспластические синдромы представляют собой гетерогенную группу клональных заболеваний системы крови, характеризующихся цитопенией, признаками дисмиелопоэза и высоким
- 54. В 80-90 % случаев этиология МДС неизвестна, в 10-15 % развитию заболевания предшествовала цитостатическая и/или лучевая
- 55. ВОЗ –классификация МДС.
- 58. Скачать презентацию

Миелопролиферативные заболевания
представляют собой клональные заболевания, возникающие на уровне стволовой кроветворной клетки,
Миелопролиферативные заболевания
представляют собой клональные заболевания, возникающие на уровне стволовой кроветворной клетки,
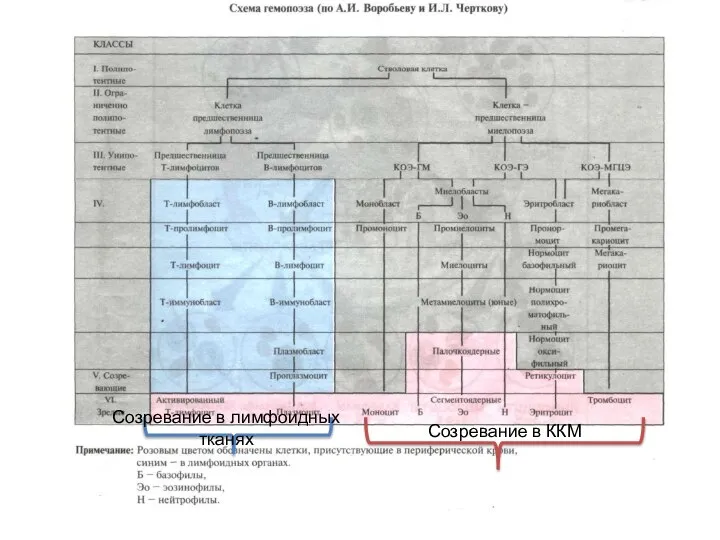
Созревание в ККМ
Созревание в лимфоидных тканях
Созревание в ККМ
Созревание в лимфоидных тканях

КЛАССИФИКАЦИЯ МИЕЛОИДНЫХ НЕОПЛАЗИЙ (ВСЕМИРНАЯ ОРГАНИЗАЦИЯ ЗДРАВООХРАНЕНИЯ, ПЕРЕСМОТР 2016)
1. Миелопролиферативные заболевания (МПЗ)
a. Хронический миелолейкоз
КЛАССИФИКАЦИЯ МИЕЛОИДНЫХ НЕОПЛАЗИЙ (ВСЕМИРНАЯ ОРГАНИЗАЦИЯ ЗДРАВООХРАНЕНИЯ, ПЕРЕСМОТР 2016)
1. Миелопролиферативные заболевания (МПЗ)
a. Хронический миелолейкоз

Хронические миелопролиферативные заболевания
1. Хронический миелоидный лейкоз (Ph+)(филадельфийская хромосома)
(Ph-):
2. Истинная полицитемия (ИП)
3.
Хронические миелопролиферативные заболевания
1. Хронический миелоидный лейкоз (Ph+)(филадельфийская хромосома)
(Ph-):
2. Истинная полицитемия (ИП)
3.

Хронический миелоидный лейкоз
Злокачественная опухоль кроветворной ткани,
исходящая из клеток-предшественниц миелопоэза,
морфологическим субстратом которой
Хронический миелоидный лейкоз
Злокачественная опухоль кроветворной ткани,
исходящая из клеток-предшественниц миелопоэза,
морфологическим субстратом которой

ХМЛ
является клональным миелопролиферативным заболеванием, развивающимся в результате злокачественной трансформации в ранних
ХМЛ
является клональным миелопролиферативным заболеванием, развивающимся в результате злокачественной трансформации в ранних
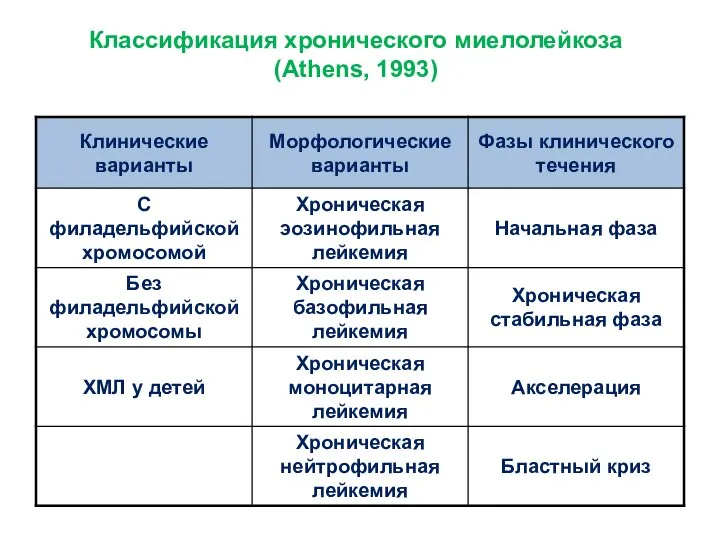
Классификация хронического миелолейкоза
(Athens, 1993)
Классификация хронического миелолейкоза
(Athens, 1993)
Хронический миелолейкоз на начальных стадиях протекает бессимптомно.
Спустя некоторое время у больных
Хронический миелолейкоз на начальных стадиях протекает бессимптомно.
Спустя некоторое время у больных
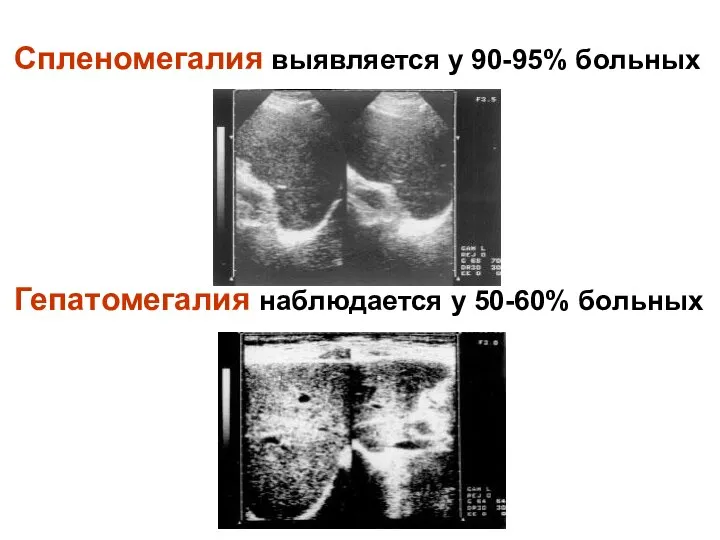
Спленомегалия выявляется у 90-95% больных
Гепатомегалия наблюдается у 50-60% больных
Спленомегалия выявляется у 90-95% больных
Гепатомегалия наблюдается у 50-60% больных

По мере накопления лейкемического клона, нарастания гепато- и спленомегалии, подавления нормального
По мере накопления лейкемического клона, нарастания гепато- и спленомегалии, подавления нормального
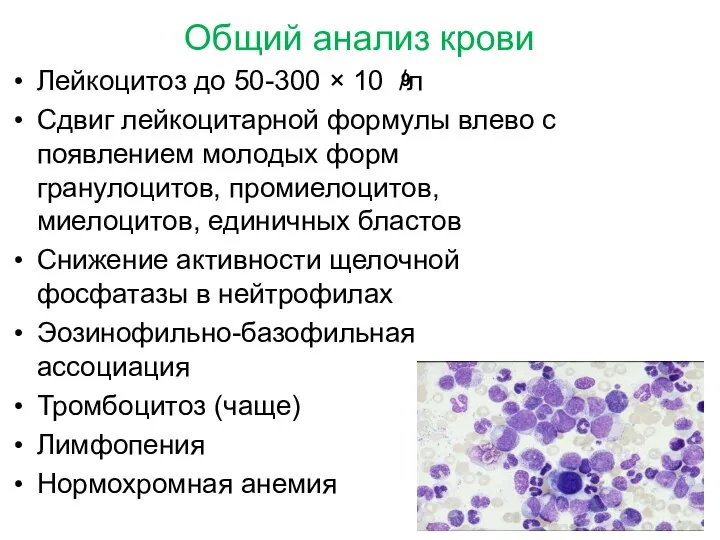
Общий анализ крови
Лейкоцитоз до 50-300 × 10 /л
Сдвиг лейкоцитарной формулы влево
Общий анализ крови
Лейкоцитоз до 50-300 × 10 /л
Сдвиг лейкоцитарной формулы влево
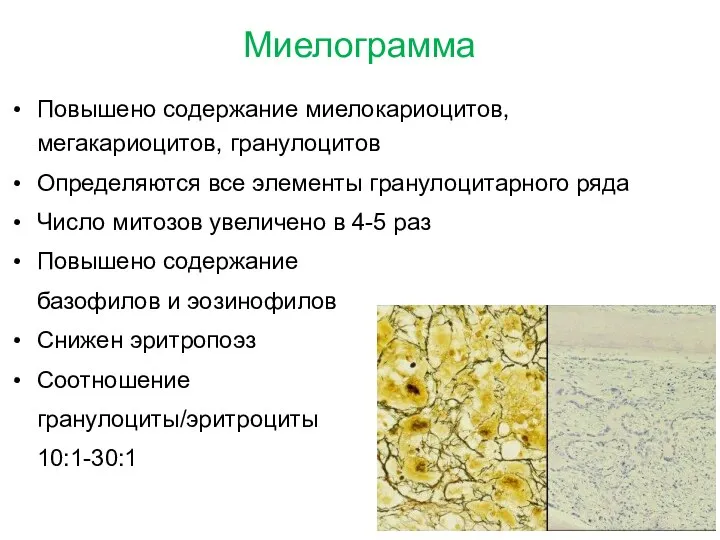
Миелограмма
Повышено содержание миелокариоцитов, мегакариоцитов, гранулоцитов
Определяются все элементы гранулоцитарного ряда
Число митозов увеличено
Миелограмма
Повышено содержание миелокариоцитов, мегакариоцитов, гранулоцитов
Определяются все элементы гранулоцитарного ряда
Число митозов увеличено

Бластный криз
Выраженная анемия
Лейкоцитоз
Нейтропения
Бластемия 30% и более
«Провал» в лейкоцитарной формуле
Тромбоцитопения
Увеличение количества базофилов
Бластный криз
Выраженная анемия
Лейкоцитоз
Нейтропения
Бластемия 30% и более
«Провал» в лейкоцитарной формуле
Тромбоцитопения
Увеличение количества базофилов

Фазы ХМЛ по классификациям ELN
Фазы ХМЛ по классификациям ELN

Цель современной терапии ХМЛ
максимальное подавление Ph-положительного опухолевого клона,
снижение риска
Цель современной терапии ХМЛ
максимальное подавление Ph-положительного опухолевого клона,
снижение риска

В период обследования, до получения результатов цитогенетического исследования, подтверждающих наличие Ph-хромосомы
В период обследования, до получения результатов цитогенетического исследования, подтверждающих наличие Ph-хромосомы

Современные направления терапии ХМЛ:
Альфа-интерферон
антагонист ростовых факторов, антипролиферативная активность, стимуляция противоопухолевого иммунитета
цитогенетические
Современные направления терапии ХМЛ:
Альфа-интерферон
антагонист ростовых факторов, антипролиферативная активность, стимуляция противоопухолевого иммунитета
цитогенетические

После подтверждения диагноза ХМЛ должна быть начата терапия ИТК (ингибиторами BCR-ABL-тирозинкиназы).
После подтверждения диагноза ХМЛ должна быть начата терапия ИТК (ингибиторами BCR-ABL-тирозинкиназы).

Аллогенная трансплантация гемопоэтических стволовых клеток (алло-ТГСК) должна быть обязательно рассмотрена для
Аллогенная трансплантация гемопоэтических стволовых клеток (алло-ТГСК) должна быть обязательно рассмотрена для

Истинная полицитемия (ИП)
син.: эритремия, болезнь Вакеза, истинная красная полицитемия --
Истинная полицитемия (ИП)
син.: эритремия, болезнь Вакеза, истинная красная полицитемия --
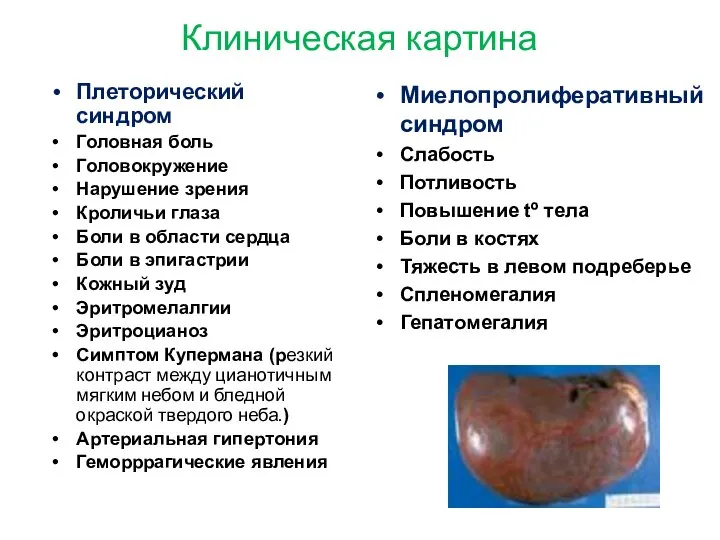
Клиническая картина
Плеторический синдром
Головная боль
Головокружение
Нарушение зрения
Кроличьи глаза
Боли в области сердца
Боли в эпигастрии
Кожный
Клиническая картина
Плеторический синдром
Головная боль
Головокружение
Нарушение зрения
Кроличьи глаза
Боли в области сердца
Боли в эпигастрии
Кожный
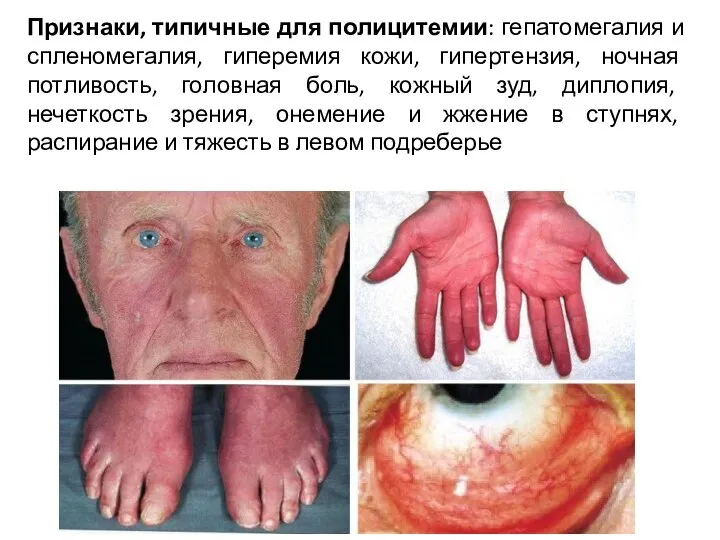
Признаки, типичные для полицитемии: гепатомегалия и спленомегалия, гиперемия кожи, гипертензия, ночная
Признаки, типичные для полицитемии: гепатомегалия и спленомегалия, гиперемия кожи, гипертензия, ночная
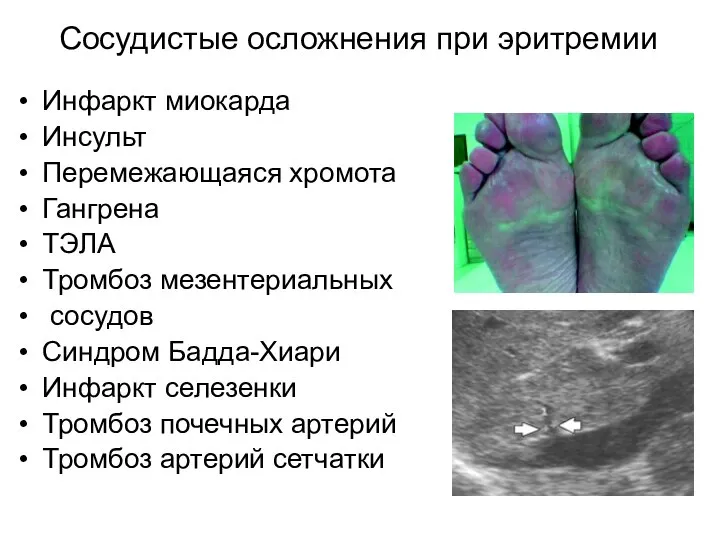
Сосудистые осложнения при эритремии
Инфаркт миокарда
Инсульт
Перемежающаяся хромота
Гангрена
ТЭЛА
Тромбоз мезентериальных
сосудов
Синдром Бадда-Хиари
Инфаркт селезенки
Тромбоз почечных
Сосудистые осложнения при эритремии
Инфаркт миокарда
Инсульт
Перемежающаяся хромота
Гангрена
ТЭЛА
Тромбоз мезентериальных
сосудов
Синдром Бадда-Хиари
Инфаркт селезенки
Тромбоз почечных
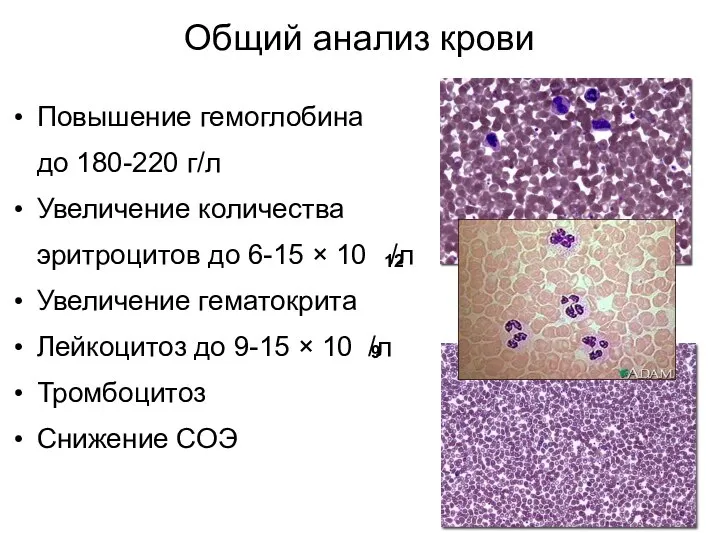
Общий анализ крови
Повышение гемоглобина
до 180-220 г/л
Увеличение количества
эритроцитов до 6-15 ×
Общий анализ крови
Повышение гемоглобина
до 180-220 г/л
Увеличение количества
эритроцитов до 6-15 ×
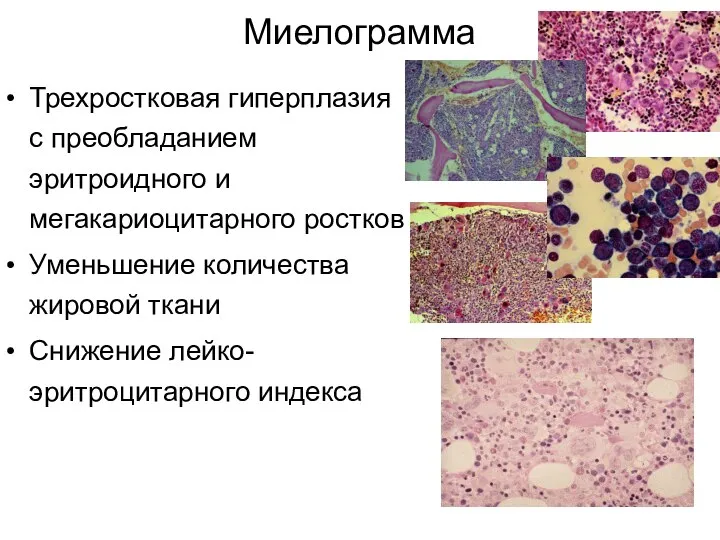
Миелограмма
Трехростковая гиперплазия с преобладанием эритроидного и мегакариоцитарного ростков
Уменьшение количества жировой ткани
Снижение
Миелограмма
Трехростковая гиперплазия с преобладанием эритроидного и мегакариоцитарного ростков
Уменьшение количества жировой ткани
Снижение

Лабораторная диагностика
Биохимический анализ крови
повышение содержания мочевой кислоты
Радиологические методы
увеличение объема циркулирующих эртроцитов,
Лабораторная диагностика
Биохимический анализ крови
повышение содержания мочевой кислоты
Радиологические методы
увеличение объема циркулирующих эртроцитов,

Стадии эритремии
I стадия – начальная
II стадия – эритремическая
II A – без
Стадии эритремии
I стадия – начальная
II стадия – эритремическая
II A – без

Критерии для диагностики истинной полицитемии
Большие критерии
Hb более 16,5 г/мл
Критерии для диагностики истинной полицитемии
Большие критерии
Hb более 16,5 г/мл

Вторичные эритроцитозы
Абсолютные
Вследствие генерализованной тканевой гипоксии
высотная болезнь, обструктивные заболевания легких, пороки сердца,
Вторичные эритроцитозы
Абсолютные
Вследствие генерализованной тканевой гипоксии
высотная болезнь, обструктивные заболевания легких, пороки сердца,

Лечение эритремии
Кровопускание
400-500 мл через день, гирудотерапия
Дезагреганты
аспирин, тиклид, трентал, реополиглюкин
Цитостатическая терапия
гидреа, миелосан,
Лечение эритремии
Кровопускание
400-500 мл через день, гирудотерапия
Дезагреганты
аспирин, тиклид, трентал, реополиглюкин
Цитостатическая терапия
гидреа, миелосан,

Суммированные рекомендации по лечению ИП
1. Для всех больных:
• кровопускания/эритроцитаферез для поддержания
Суммированные рекомендации по лечению ИП
1. Для всех больных:
• кровопускания/эритроцитаферез для поддержания

2. Для больных группы низкого риска развития тромбогеморрагических осложнений.
• Циторедуктивная терапия
2. Для больных группы низкого риска развития тромбогеморрагических осложнений.
• Циторедуктивная терапия

Эссенциальная тромбоцитемия (ЭТ)
(син.: первичный тромбоцитоз, идиопатический тромбоцитоз, геморрагическая тромбоцитемия) – клональное
Эссенциальная тромбоцитемия (ЭТ)
(син.: первичный тромбоцитоз, идиопатический тромбоцитоз, геморрагическая тромбоцитемия) – клональное

Критерии диагностики эссенциальной тромбоцитемии
Большие критерии
Количество тромбоцитов 450·109 /л и более.
Критерии диагностики эссенциальной тромбоцитемии
Большие критерии
Количество тромбоцитов 450·109 /л и более.

Эссенциальная тромбоцитемия
проявляется обмороками и предобморочными состояниями, болью в груди, пульсирующей боль
Эссенциальная тромбоцитемия
проявляется обмороками и предобморочными состояниями, болью в груди, пульсирующей боль

Суммированные рекомендации при ЭТ
1 Для всех больных:
-- профилактика сердечно-сосудистых заболеваний (устранение
Суммированные рекомендации при ЭТ
1 Для всех больных:
-- профилактика сердечно-сосудистых заболеваний (устранение

Сублейкемический миелоз (идиопатический миелофиброз)
Хроническое клональное миелопролиферативное
заболевание, развивающееся вследствие
нарушений стволовой кроветворной клетки
и
Сублейкемический миелоз (идиопатический миелофиброз)
Хроническое клональное миелопролиферативное
заболевание, развивающееся вследствие
нарушений стволовой кроветворной клетки
и

Критерии диагностики первичного миелофиброза
Критерии диагностики первичного миелофиброза
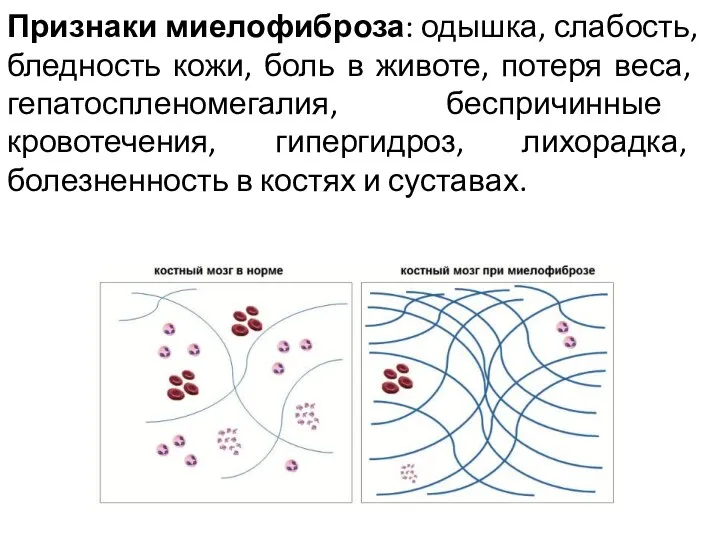
Признаки миелофиброза: одышка, слабость, бледность кожи, боль в животе, потеря веса,
Признаки миелофиброза: одышка, слабость, бледность кожи, боль в животе, потеря веса,

Клиническая картина
общая слабость
потливость
снижение аппетита
похудание
боли в костях
тяжесть и боли в подреберьях
снижение слуха
повышение
Клиническая картина
общая слабость
потливость
снижение аппетита
похудание
боли в костях
тяжесть и боли в подреберьях
снижение слуха
повышение
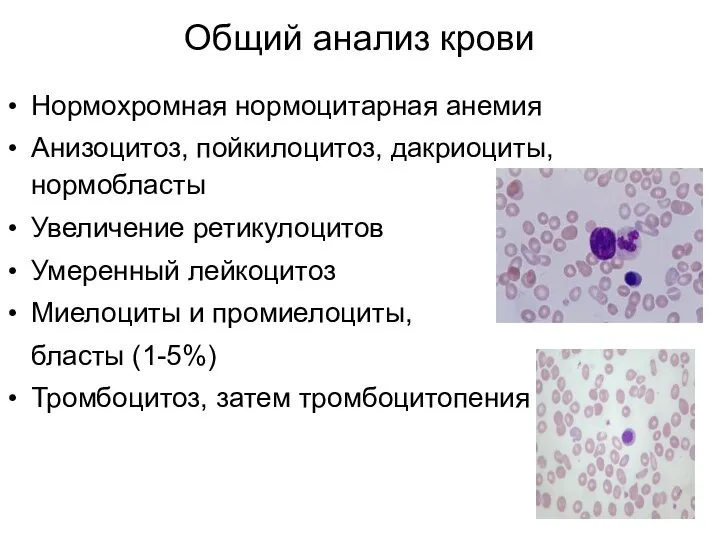
Общий анализ крови
Нормохромная нормоцитарная анемия
Анизоцитоз, пойкилоцитоз, дакриоциты, нормобласты
Увеличение ретикулоцитов
Умеренный лейкоцитоз
Миелоциты и
Общий анализ крови
Нормохромная нормоцитарная анемия
Анизоцитоз, пойкилоцитоз, дакриоциты, нормобласты
Увеличение ретикулоцитов
Умеренный лейкоцитоз
Миелоциты и
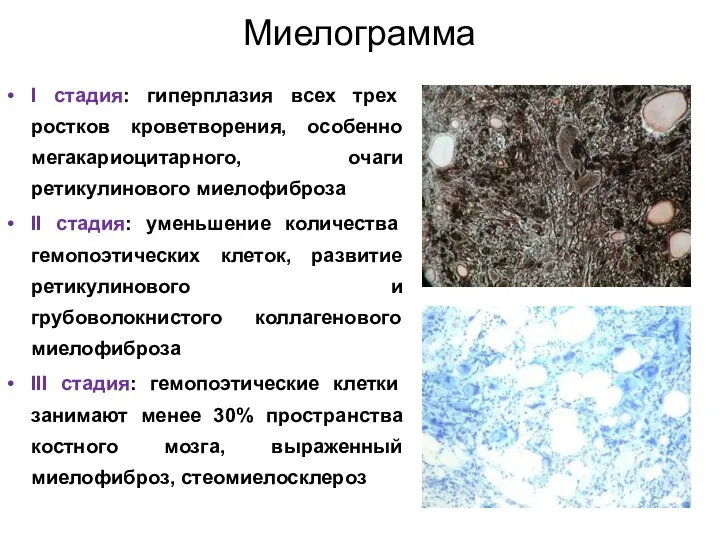
Миелограмма
I стадия: гиперплазия всех трех ростков кроветворения, особенно мегакариоцитарного, очаги ретикулинового
Миелограмма
I стадия: гиперплазия всех трех ростков кроветворения, особенно мегакариоцитарного, очаги ретикулинового

В пересмотренной Классификации ВОЗ (2017) подчеркивается важность дифференциальной диагностики эссенциальной тромбоцитемии
В пересмотренной Классификации ВОЗ (2017) подчеркивается важность дифференциальной диагностики эссенциальной тромбоцитемии

Методы терапии ПМФ
Алло-ТГСК;
Медикаментозная терапия : цитостатики, эритропоэзстимулирующие агенты, глюкокортикостероиды, андрогены, ингибиторы
Методы терапии ПМФ
Алло-ТГСК;
Медикаментозная терапия : цитостатики, эритропоэзстимулирующие агенты, глюкокортикостероиды, андрогены, ингибиторы

Осложнения при ПМФ и тактика их лечения
Опухолевая интоксикация
Спленомегалия
Анемия
Инфекционные осложнения (Лейкопения,нейтропения)
Тромбоцитопения и
Осложнения при ПМФ и тактика их лечения
Опухолевая интоксикация
Спленомегалия
Анемия
Инфекционные осложнения (Лейкопения,нейтропения)
Тромбоцитопения и

Бластная фаза МПЗ является терминальной стадией, прогноз неблагоприятный -- средняя продолжительность
Бластная фаза МПЗ является терминальной стадией, прогноз неблагоприятный -- средняя продолжительность

Прогноз
Хронический миелолейкоз
продолжительность жизни в среднем 3-5 лет, у отдельных больных 7-8
Прогноз
Хронический миелолейкоз
продолжительность жизни в среднем 3-5 лет, у отдельных больных 7-8

«Миелопролиферативное заболевание, неклассифицируемое»
Нозологическая форма «Миелопролиферативное заболевание, неклассифицируемое» устанавливается при наличии несомненных
«Миелопролиферативное заболевание, неклассифицируемое»
Нозологическая форма «Миелопролиферативное заболевание, неклассифицируемое» устанавливается при наличии несомненных


Мастоцитозы
В новой редакции ВОЗ (2017) группа мастоцитозов выведена из группы МПЗ
Мастоцитозы
В новой редакции ВОЗ (2017) группа мастоцитозов выведена из группы МПЗ

КЛАССИФИКАЦИЯ МИЕЛОИДНЫХ НЕОПЛАЗИЙ (ВСЕМИРНАЯ ОРГАНИЗАЦИЯ ЗДРАВООХРАНЕНИЯ, ПЕРЕСМОТР 2016)
1. Миелопролиферативные заболевания (МПЗ)
a. Хронический миелолейкоз
КЛАССИФИКАЦИЯ МИЕЛОИДНЫХ НЕОПЛАЗИЙ (ВСЕМИРНАЯ ОРГАНИЗАЦИЯ ЗДРАВООХРАНЕНИЯ, ПЕРЕСМОТР 2016)
1. Миелопролиферативные заболевания (МПЗ)
a. Хронический миелолейкоз

Миелодиспластические синдромы
представляют собой гетерогенную группу клональных заболеваний системы крови, характеризующихся цитопенией,
Миелодиспластические синдромы
представляют собой гетерогенную группу клональных заболеваний системы крови, характеризующихся цитопенией,

В 80-90 % случаев этиология МДС неизвестна,
в 10-15 % развитию заболевания
В 80-90 % случаев этиология МДС неизвестна,
в 10-15 % развитию заболевания

ВОЗ –классификация МДС.
ВОЗ –классификация МДС.

 Миома матки
Миома матки Классификация антиадренергических средств
Классификация антиадренергических средств Здоровое питание
Здоровое питание Hysterosalpingography (HSG)
Hysterosalpingography (HSG) Болезнь Лайма
Болезнь Лайма Ортопедические методы лечения переломов нижней челюстей репонирующими аппаратами
Ортопедические методы лечения переломов нижней челюстей репонирующими аппаратами Расстройства кровообращения (артериальное полнокровие, венозное полнокровие)
Расстройства кровообращения (артериальное полнокровие, венозное полнокровие) Глаукома нормального давления
Глаукома нормального давления Ринологиядағы синдромдар
Ринологиядағы синдромдар Организационная психология
Организационная психология Тема 3. Конддрашкина. Обструкция и гипердилатация желчного протока
Тема 3. Конддрашкина. Обструкция и гипердилатация желчного протока Классическая школа управления. Школа научного менеджмента. Административная школа управления
Классическая школа управления. Школа научного менеджмента. Административная школа управления Йога – учение о познании себя и окружающего мира
Йога – учение о познании себя и окружающего мира Лечебно-профилактическое питание
Лечебно-профилактическое питание Проблемы климактерия
Проблемы климактерия Роговица глаза. Аномалии развития роговицы
Роговица глаза. Аномалии развития роговицы Печеночная недостаточность
Печеночная недостаточность Стационарная реабилитация родителей, страдающих наркологическими расстройствами воспитывающих несовершеннолетних детей
Стационарная реабилитация родителей, страдающих наркологическими расстройствами воспитывающих несовершеннолетних детей Заболевания пищевода. Смещения пищевода. Дивертикулы. Инородные тела. Ожоги. (Часть 2)
Заболевания пищевода. Смещения пищевода. Дивертикулы. Инородные тела. Ожоги. (Часть 2) Системная красная волчанка
Системная красная волчанка Современное состояние и перспективы развития иммунопрофилактики в ВС РФ
Современное состояние и перспективы развития иммунопрофилактики в ВС РФ Общие основы физиотерапии
Общие основы физиотерапии Терапиялық стоматология. Түбір өзектерді обтурациялайтын материалдар (силерлер). Жүйесі. Пластикалық қатаятын материалдар
Терапиялық стоматология. Түбір өзектерді обтурациялайтын материалдар (силерлер). Жүйесі. Пластикалық қатаятын материалдар Аденовирусы. Симптомы аденовирусной инфекции, диагностика, лечение
Аденовирусы. Симптомы аденовирусной инфекции, диагностика, лечение Антигипертензивные или гипотензивные лекарственные средства
Антигипертензивные или гипотензивные лекарственные средства Профилактика инсульта
Профилактика инсульта Стомы. Виды стом
Стомы. Виды стом Фронтальная ТРГ
Фронтальная ТРГ