- Мукополисахаридозы

Содержание
- 2. МУКОПОЛИСАХАРИДОЗЫ (МУКОПОЛИСАХАРИДЫ + -ŌSIS) группа наследственных болезней соединительной ткани, обусловленных нарушением обмена гликозаминогликанов (кислых мукополисахаридов) в
- 3. МУКОПОЛИСАХАРИДОЗ ТИПА I-Н (СИНДРОМ ГУРЛЕР). Впервые описан немецким педиатром Гурлер (G. Hurler) в 1919 г. Часто
- 4. Синдром Гурлер: типичные внешние проявления.
- 5. АНОМАЛИИ ЛИЦЕВОГО СКЕЛЕТА ПРИ СИНДРОМЕ HURLER
- 6. ПОМУТНЕНИЕ РОГОВИЦЫ ПРИ СИНДРОМЕ HURLER МУКОПОЛИСАХАРИДОЗЫ ГРУППА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ, ОБУСЛОВЛЕННЫХ ДЕФИЦИТОМ ФЕРМЕНТОВ ГИДРОЛИЗА МУКОПОЛИСАХАРИДОВ (ГЛЮКОЗИДАЗЫ). ПРОДУКТЫ
- 7. При рентгенологическом исследовании выявляются характерные изменения позвонков и позвоночного столба (тела позвонков кубовидные с закругленными контурами,
- 8. РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА ГУРЛЕР — ДЕФОРМАЦИЯ ТАЗА И БЕДРЕННЫХ КОСТЕЙ.
- 9. РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА ГУРЛЕР — ХАРАКТЕРНЫЙ ВИД КИСТЕЙ.
- 10. МУКОПОЛИСАХАРИДОЗ ТИПА I-S (БОЛЕЗНЬ ШЕЙЕ; ПОЗДНИЙ СИНДРОМ ГУРЛЕР). Впервые описан американским офтальмологом Шейе (Н.G. Scheie) в
- 11. БОЛЕЗНЬ ШЕЙЕ: ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ
- 12. МУКОПОЛИСАХАРИДОЗ ТИПА II (СИНДРОМ ГУНТЕРА). Клинические симптомы появляются позднее, чем при синдроме Гурлер (у детей старше
- 13. СИНДРОМ ГУНТЕРА У МАЛЬЧИКА 2 ЛЕТ — ГРУБЫЕ ЧЕРТЫ ЛИЦА, СКАФОЦЕФАЛИЯ.
- 14. СИНДРОМ ГУНТЕРА У МАЛЬЧИКА 2 ЛЕТ — ИЗМЕНЕНИЯ СКЕЛЕТА СЛАБО ВЫРАЖЕНЫ, НЕТ КИФОЗА, КОНТРАКТУР.
- 15. МУКОПОЛИСАХАРИДОЗ ТИПА III (СИНДРОМ САНФИЛИППО, БОЛЕЗНЬ САНФИЛИППО). Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в 1963 г.
- 16. МУКОПОЛИСАХАРИДОЗ ТИПА IV (СНИДРОМ МОРКИО, БОЛЕЗНЬ МОРКИО). Заболевание в 1929 г. независимо друг от друга впервые
- 17. СИНДРОМ МОРКИО: ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ.
- 18. РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ИСКРИВЛЕНИЕ ПОЗВОНОЧНИКА И ПЛАТИСПОНДИЛИЯ.
- 19. РИС. 6Б). РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ДЕФОРМАЦИЯ КОСТЕЙ ТАЗА, ВЕРТЛУЖНЫХ ВПАДИН, ГИПОПЛАЗИЯ ГОЛОВОК БЕДРЕННЫХ КОСТЕЙ.
- 20. РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ДЕФОРМАЦИЯ КОСТЕЙ КИСТИ.
- 21. МУКОПОЛИСАХАРИДОЗ ТИПА VI (СИНДРОМ МАРОТО — ЛАМИ, БОЛЕЗНЬ МАРОТО — ЛАМИ) в 1960 г. впервые описан
- 22. СИНДРОМ МАРОТО — ЛАМИ: (ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ У ДЕВОЧКИ 9 ЛЕТ) — ГРУБЫЕ ЧЕРТЫ ЛИЦА, БОЧКООБРАЗНАЯ
- 23. СИНДРОМ МАРОТО — ЛАМИ: (ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ У ДЕВОЧКИ 9 ЛЕТ) — КОНТРАКТУРЫ ВЕРХНИХ И НИЖНИХ
- 24. РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МАРОТО — ЛАМИ — ИЗМЕНЕНИЯ ПОЗВОНОЧНИКА.
- 25. РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МАРОТО — ЛАМИ — ДЕФОРМАЦИЯ КОСТЕЙ ТАЗА.
- 26. СИНДРОМ МОРКИО-УЛЛЬРИХА. ГЕНЕТИЧЕСКИ ДЕТЕРМИНИРОВАННОЕ ЗАБОЛЕВАНИЕ - МУКОПОЛИСАХАРИДОЗ - IV. ХОНДРОДИСТРОФИЯ ХАРАКТЕРИЗУЕТСЯ ОСЛАБЛЕНИЕМ СВЯЗОЧНОГО АППАРАТА СУСТАВОВ, ЗНАЧИТЕЛЬНЫМИ
- 27. РЕДКО ВСТРЕЧАЕМЫЕ ТИПЫ МУКОПОЛИСАХАРИДОЗОВ Мукополисахаридоз типа VII (синдром Слая). Описан Слаем (W.S. Sly) в 1973 г.
- 28. ДИАГНОЗ ОСНОВЫВАЕТСЯ НА КЛИНИЧЕСКИХ ПРОЯВЛЕНИЯХ, ДАННЫХ РЕНТГЕНОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ, ОПРЕДЕЛЕНИИ ЭКСКРЕЦИИ С МОЧОЙ ГЛИКОЗАМИНОГЛИКАНОВ, ИССЛЕДОВАНИИ АКТИВНОСТИ СПЕЦИФИЧЕСКИХ
- 30. Скачать презентацию

МУКОПОЛИСАХАРИДОЗЫ
(МУКОПОЛИСАХАРИДЫ + -ŌSIS)
группа наследственных болезней соединительной ткани, обусловленных нарушением обмена
МУКОПОЛИСАХАРИДОЗЫ
(МУКОПОЛИСАХАРИДЫ + -ŌSIS)
группа наследственных болезней соединительной ткани, обусловленных нарушением обмена

МУКОПОЛИСАХАРИДОЗ ТИПА I-Н (СИНДРОМ ГУРЛЕР).
Впервые описан немецким педиатром Гурлер (G. Hurler) в
МУКОПОЛИСАХАРИДОЗ ТИПА I-Н (СИНДРОМ ГУРЛЕР).
Впервые описан немецким педиатром Гурлер (G. Hurler) в
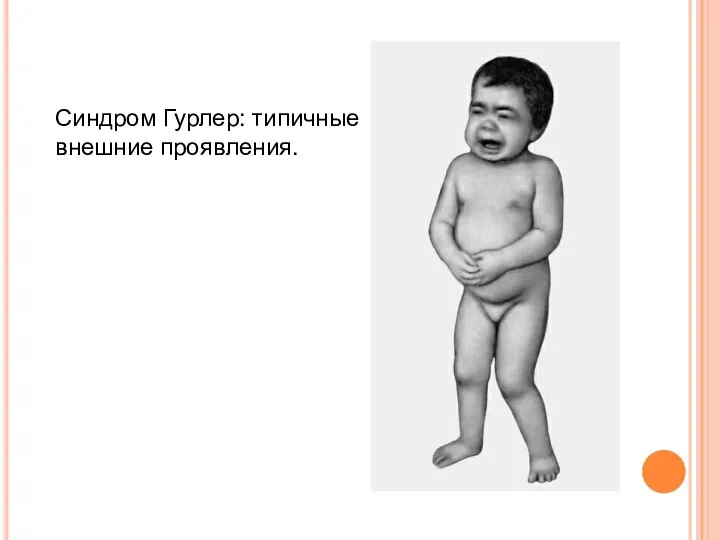
Синдром Гурлер: типичные внешние проявления.
Синдром Гурлер: типичные внешние проявления.
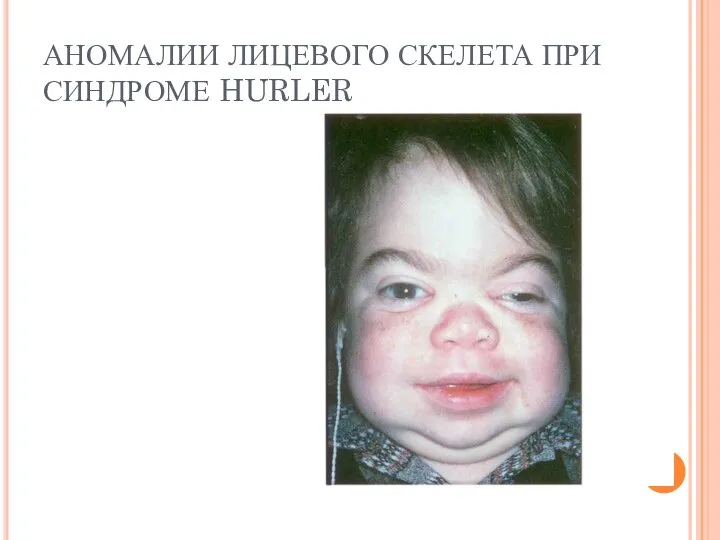
АНОМАЛИИ ЛИЦЕВОГО СКЕЛЕТА ПРИ СИНДРОМЕ HURLER
АНОМАЛИИ ЛИЦЕВОГО СКЕЛЕТА ПРИ СИНДРОМЕ HURLER
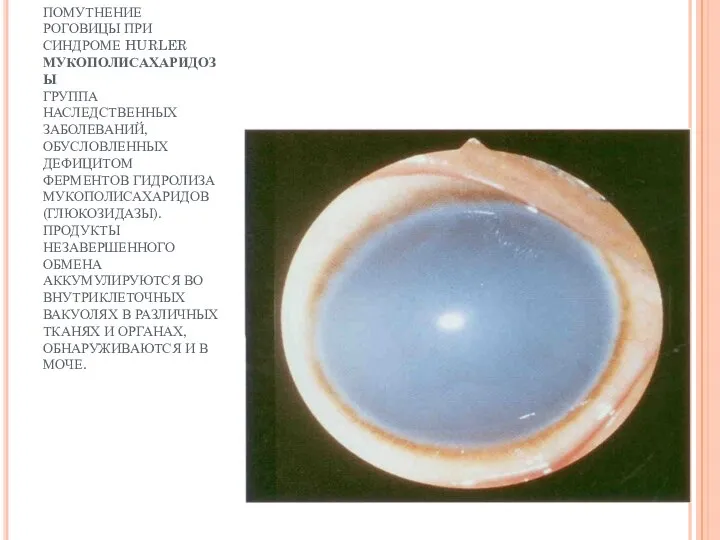
ПОМУТНЕНИЕ РОГОВИЦЫ ПРИ СИНДРОМЕ HURLER
МУКОПОЛИСАХАРИДОЗЫ
ГРУППА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ, ОБУСЛОВЛЕННЫХ ДЕФИЦИТОМ ФЕРМЕНТОВ
ПОМУТНЕНИЕ РОГОВИЦЫ ПРИ СИНДРОМЕ HURLER МУКОПОЛИСАХАРИДОЗЫ ГРУППА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ, ОБУСЛОВЛЕННЫХ ДЕФИЦИТОМ ФЕРМЕНТОВ

При рентгенологическом исследовании выявляются характерные изменения позвонков и позвоночного столба (тела
При рентгенологическом исследовании выявляются характерные изменения позвонков и позвоночного столба (тела
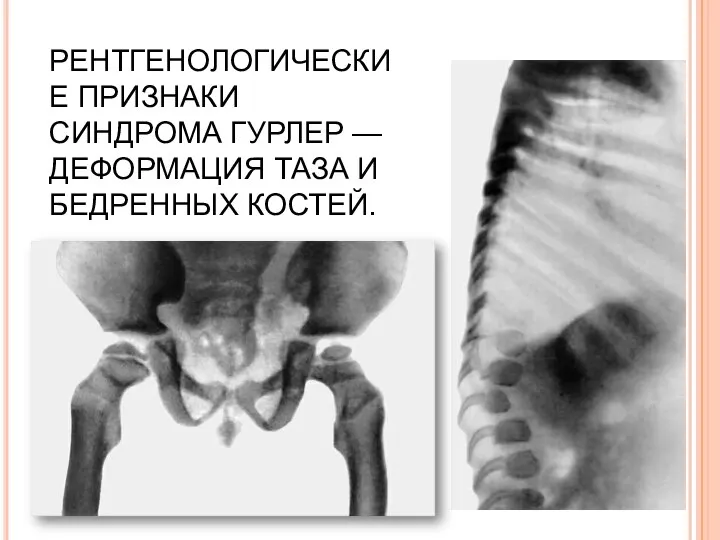
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА ГУРЛЕР — ДЕФОРМАЦИЯ ТАЗА И БЕДРЕННЫХ КОСТЕЙ.
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА ГУРЛЕР — ДЕФОРМАЦИЯ ТАЗА И БЕДРЕННЫХ КОСТЕЙ.
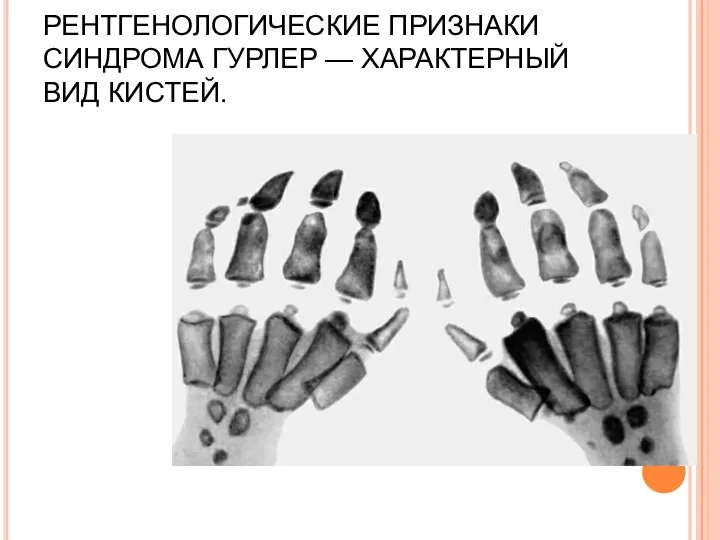
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА ГУРЛЕР — ХАРАКТЕРНЫЙ ВИД КИСТЕЙ.
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА ГУРЛЕР — ХАРАКТЕРНЫЙ ВИД КИСТЕЙ.

МУКОПОЛИСАХАРИДОЗ ТИПА I-S (БОЛЕЗНЬ ШЕЙЕ; ПОЗДНИЙ СИНДРОМ ГУРЛЕР).
Впервые описан американским офтальмологом
МУКОПОЛИСАХАРИДОЗ ТИПА I-S (БОЛЕЗНЬ ШЕЙЕ; ПОЗДНИЙ СИНДРОМ ГУРЛЕР).
Впервые описан американским офтальмологом
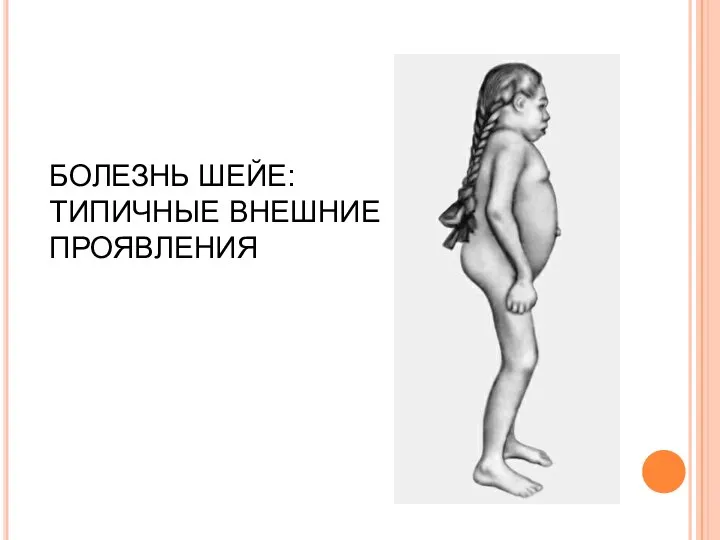
БОЛЕЗНЬ ШЕЙЕ: ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ
БОЛЕЗНЬ ШЕЙЕ: ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ

МУКОПОЛИСАХАРИДОЗ ТИПА II (СИНДРОМ ГУНТЕРА).
Клинические симптомы появляются позднее, чем при синдроме
МУКОПОЛИСАХАРИДОЗ ТИПА II (СИНДРОМ ГУНТЕРА).
Клинические симптомы появляются позднее, чем при синдроме
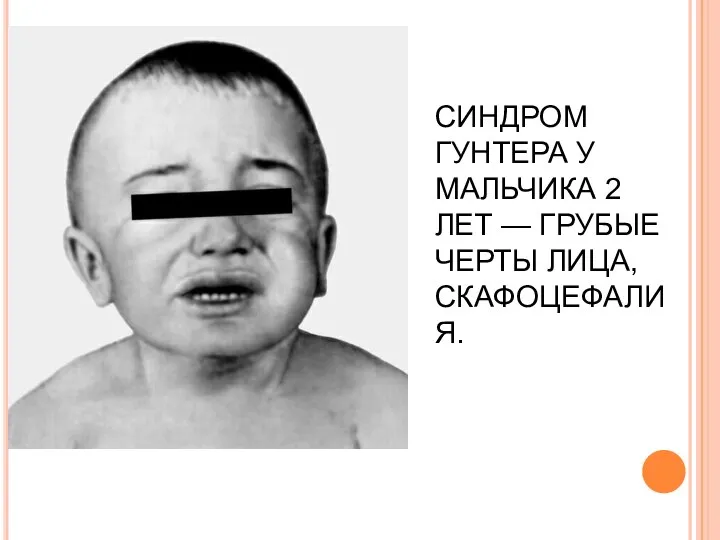
СИНДРОМ ГУНТЕРА У МАЛЬЧИКА 2 ЛЕТ — ГРУБЫЕ ЧЕРТЫ ЛИЦА, СКАФОЦЕФАЛИЯ.
СИНДРОМ ГУНТЕРА У МАЛЬЧИКА 2 ЛЕТ — ГРУБЫЕ ЧЕРТЫ ЛИЦА, СКАФОЦЕФАЛИЯ.

СИНДРОМ ГУНТЕРА У МАЛЬЧИКА 2 ЛЕТ — ИЗМЕНЕНИЯ СКЕЛЕТА СЛАБО ВЫРАЖЕНЫ, НЕТ
СИНДРОМ ГУНТЕРА У МАЛЬЧИКА 2 ЛЕТ — ИЗМЕНЕНИЯ СКЕЛЕТА СЛАБО ВЫРАЖЕНЫ, НЕТ

МУКОПОЛИСАХАРИДОЗ ТИПА III (СИНДРОМ САНФИЛИППО, БОЛЕЗНЬ САНФИЛИППО).
Описан американским педиатром Санфилиппо (S.J. Sanfilippo)
МУКОПОЛИСАХАРИДОЗ ТИПА III (СИНДРОМ САНФИЛИППО, БОЛЕЗНЬ САНФИЛИППО).
Описан американским педиатром Санфилиппо (S.J. Sanfilippo)

МУКОПОЛИСАХАРИДОЗ ТИПА IV (СНИДРОМ МОРКИО, БОЛЕЗНЬ МОРКИО).
Заболевание в 1929 г. независимо друг
МУКОПОЛИСАХАРИДОЗ ТИПА IV (СНИДРОМ МОРКИО, БОЛЕЗНЬ МОРКИО).
Заболевание в 1929 г. независимо друг
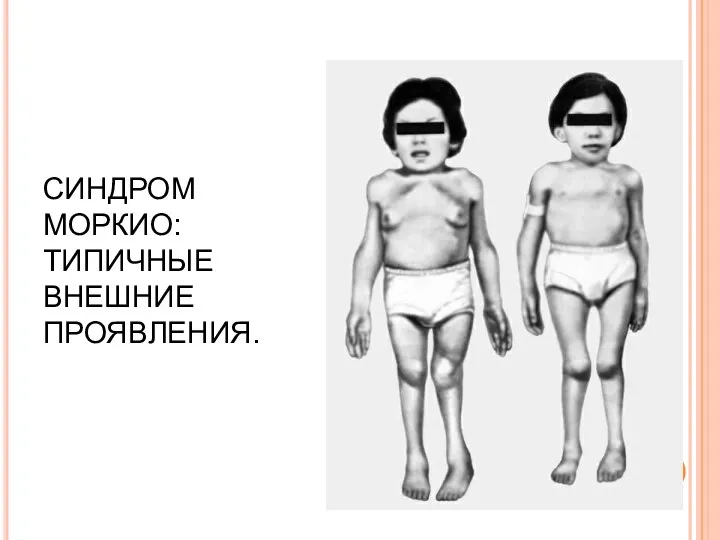
СИНДРОМ МОРКИО: ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ.
СИНДРОМ МОРКИО: ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ.
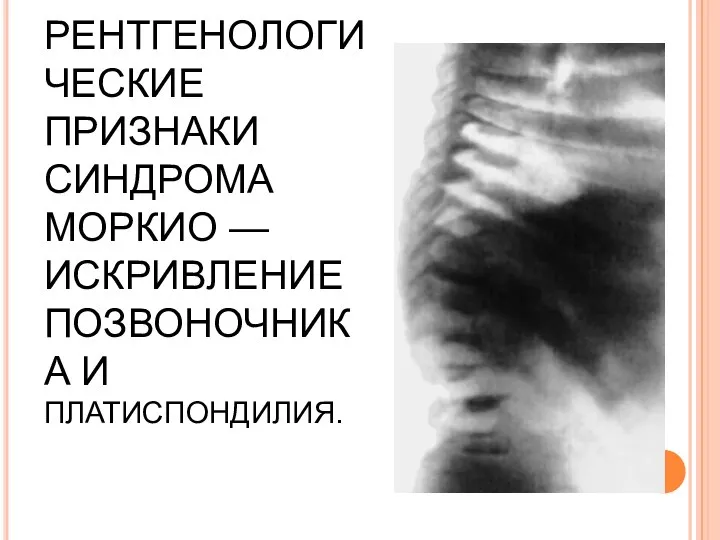
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ИСКРИВЛЕНИЕ ПОЗВОНОЧНИКА И ПЛАТИСПОНДИЛИЯ.
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ИСКРИВЛЕНИЕ ПОЗВОНОЧНИКА И ПЛАТИСПОНДИЛИЯ.
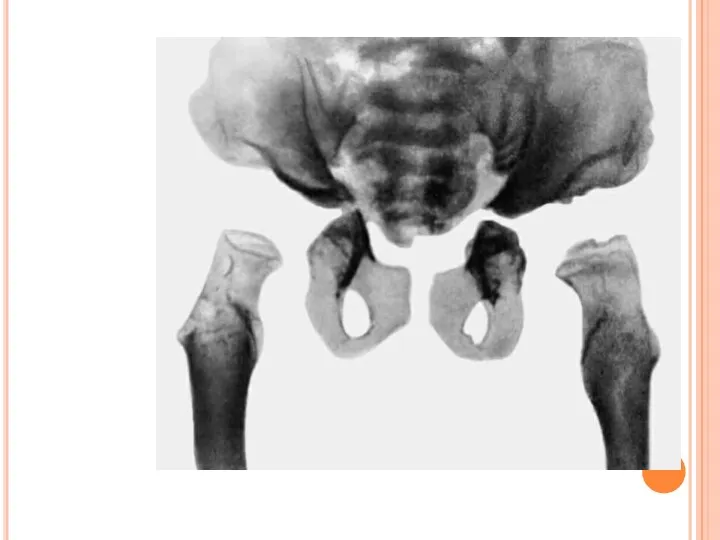
РИС. 6Б). РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ДЕФОРМАЦИЯ КОСТЕЙ ТАЗА, ВЕРТЛУЖНЫХ ВПАДИН,
РИС. 6Б). РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ДЕФОРМАЦИЯ КОСТЕЙ ТАЗА, ВЕРТЛУЖНЫХ ВПАДИН,
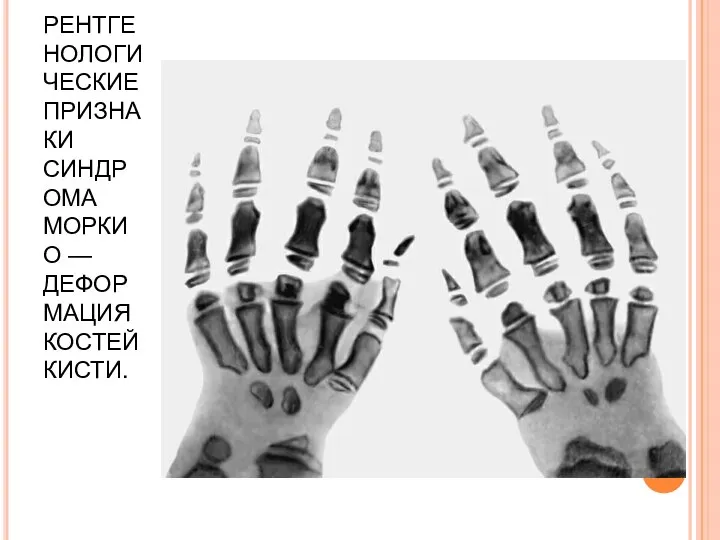
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ДЕФОРМАЦИЯ КОСТЕЙ КИСТИ.
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МОРКИО — ДЕФОРМАЦИЯ КОСТЕЙ КИСТИ.

МУКОПОЛИСАХАРИДОЗ ТИПА VI (СИНДРОМ МАРОТО — ЛАМИ, БОЛЕЗНЬ МАРОТО — ЛАМИ)
в 1960 г. впервые
МУКОПОЛИСАХАРИДОЗ ТИПА VI (СИНДРОМ МАРОТО — ЛАМИ, БОЛЕЗНЬ МАРОТО — ЛАМИ)
в 1960 г. впервые
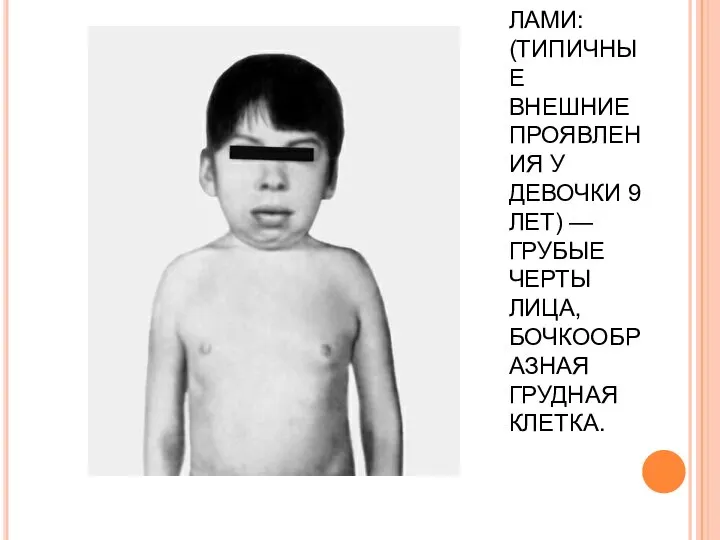
СИНДРОМ МАРОТО — ЛАМИ: (ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ У ДЕВОЧКИ 9 ЛЕТ) — ГРУБЫЕ
СИНДРОМ МАРОТО — ЛАМИ: (ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ У ДЕВОЧКИ 9 ЛЕТ) — ГРУБЫЕ

СИНДРОМ МАРОТО — ЛАМИ: (ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ У ДЕВОЧКИ 9 ЛЕТ) — КОНТРАКТУРЫ
СИНДРОМ МАРОТО — ЛАМИ: (ТИПИЧНЫЕ ВНЕШНИЕ ПРОЯВЛЕНИЯ У ДЕВОЧКИ 9 ЛЕТ) — КОНТРАКТУРЫ
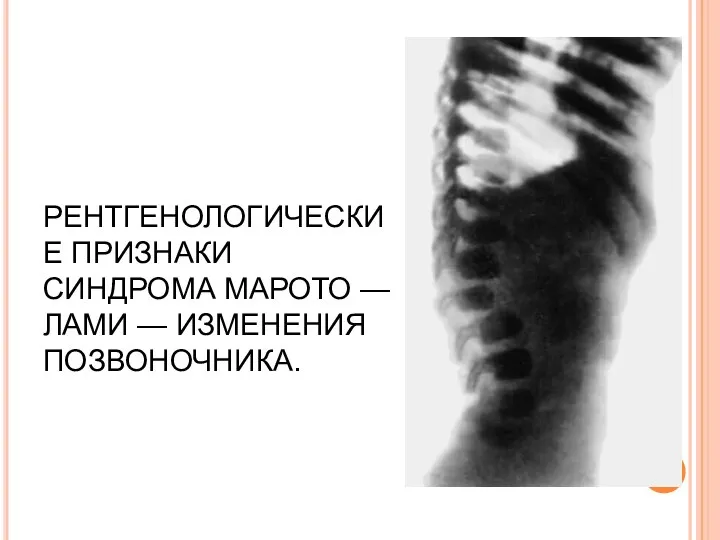
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МАРОТО — ЛАМИ — ИЗМЕНЕНИЯ ПОЗВОНОЧНИКА.
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МАРОТО — ЛАМИ — ИЗМЕНЕНИЯ ПОЗВОНОЧНИКА.
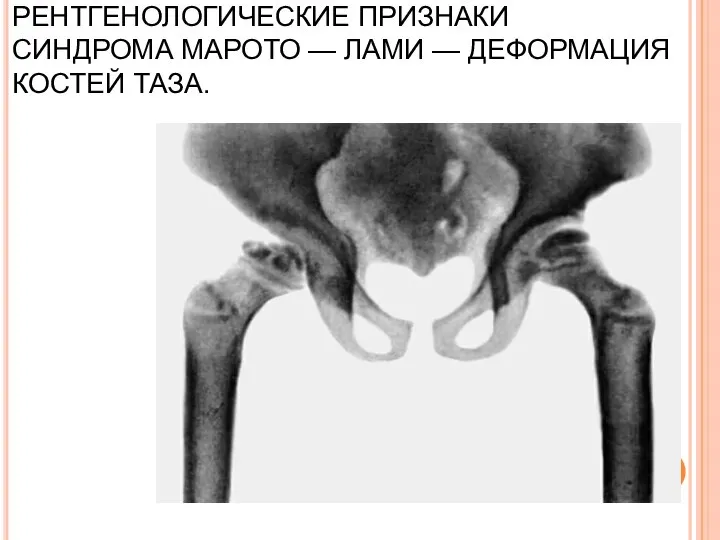
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МАРОТО — ЛАМИ — ДЕФОРМАЦИЯ КОСТЕЙ ТАЗА.
РЕНТГЕНОЛОГИЧЕСКИЕ ПРИЗНАКИ СИНДРОМА МАРОТО — ЛАМИ — ДЕФОРМАЦИЯ КОСТЕЙ ТАЗА.
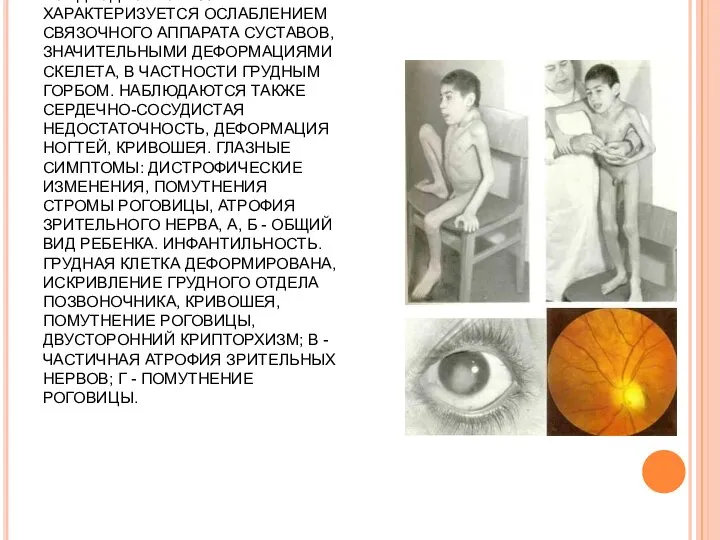
СИНДРОМ МОРКИО-УЛЛЬРИХА. ГЕНЕТИЧЕСКИ ДЕТЕРМИНИРОВАННОЕ ЗАБОЛЕВАНИЕ - МУКОПОЛИСАХАРИДОЗ - IV. ХОНДРОДИСТРОФИЯ ХАРАКТЕРИЗУЕТСЯ
СИНДРОМ МОРКИО-УЛЛЬРИХА. ГЕНЕТИЧЕСКИ ДЕТЕРМИНИРОВАННОЕ ЗАБОЛЕВАНИЕ - МУКОПОЛИСАХАРИДОЗ - IV. ХОНДРОДИСТРОФИЯ ХАРАКТЕРИЗУЕТСЯ

РЕДКО ВСТРЕЧАЕМЫЕ ТИПЫ МУКОПОЛИСАХАРИДОЗОВ
Мукополисахаридоз типа VII (синдром Слая). Описан Слаем (W.S. Sly)
РЕДКО ВСТРЕЧАЕМЫЕ ТИПЫ МУКОПОЛИСАХАРИДОЗОВ
Мукополисахаридоз типа VII (синдром Слая). Описан Слаем (W.S. Sly)

ДИАГНОЗ ОСНОВЫВАЕТСЯ НА КЛИНИЧЕСКИХ ПРОЯВЛЕНИЯХ, ДАННЫХ РЕНТГЕНОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ, ОПРЕДЕЛЕНИИ ЭКСКРЕЦИИ С
ДИАГНОЗ ОСНОВЫВАЕТСЯ НА КЛИНИЧЕСКИХ ПРОЯВЛЕНИЯХ, ДАННЫХ РЕНТГЕНОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ, ОПРЕДЕЛЕНИИ ЭКСКРЕЦИИ С
 3 главных буквы или ПМП в детском лагере
3 главных буквы или ПМП в детском лагере Клінічні прояви психічних захворювань і їх судово-психіатрична оцінка. Тема 3
Клінічні прояви психічних захворювань і їх судово-психіатрична оцінка. Тема 3 ЛР и ЛРС содержащие БАВ
ЛР и ЛРС содержащие БАВ Клиника и ведение родов при аномалиях сократительной деятельности матки
Клиника и ведение родов при аномалиях сократительной деятельности матки Судороги. Первая помощь
Судороги. Первая помощь Пневмосклероз
Пневмосклероз Воспаление
Воспаление Зачем нам нужно стекло
Зачем нам нужно стекло Медико-биологические и социальные основы здоровья
Медико-биологические и социальные основы здоровья Хронический гастрит. Язвенная болезнь
Хронический гастрит. Язвенная болезнь Компенсаторноприспособительные процессы
Компенсаторноприспособительные процессы Өндірістік химиялық удан туындаған кәсіптік аурулар
Өндірістік химиялық удан туындаған кәсіптік аурулар Скринирующие программы. Цели и задачи скринирующих программ
Скринирующие программы. Цели и задачи скринирующих программ Строение и функции почек
Строение и функции почек Вирусный гепатит C
Вирусный гепатит C Иммунитет
Иммунитет Сестринский процесс при остром панкреатите
Сестринский процесс при остром панкреатите Язвенная болезнь
Язвенная болезнь К. Хорни: изучение внутренних конфликтов человека или культурная психопатология
К. Хорни: изучение внутренних конфликтов человека или культурная психопатология Причины преждевременных родов. Классификация. Дифференциальная диагностика незрелости, недоношенности, ЗБУР плода
Причины преждевременных родов. Классификация. Дифференциальная диагностика незрелости, недоношенности, ЗБУР плода Нефроптоз
Нефроптоз Диагностика редких вариантов острых лейкозов методом проточной цитометрии
Диагностика редких вариантов острых лейкозов методом проточной цитометрии Ревматические заболевания. Макропрепараты
Ревматические заболевания. Макропрепараты Эпилепсия. Что делать при приступах эпилепсии
Эпилепсия. Что делать при приступах эпилепсии Бронхиальная астма
Бронхиальная астма Тікелей емес адреномиметиктер
Тікелей емес адреномиметиктер Болезни печени
Болезни печени Перефкрическая сенсо-моторная нейропатия у больных сахарным диабетом 2 типа
Перефкрическая сенсо-моторная нейропатия у больных сахарным диабетом 2 типа