- Наследственные болезни человека

Содержание
- 2. Генные (в основе патологические изменения структуры генов) – 8-10 % смертей до 5 лет; 2. Хромосомные
- 3. Генные заболевания 1. Чаще моногенные, т.е. в основе патологии изменение одной пары аллельных генов; 2. Большинство
- 4. Классификация генных болезней (ВОЗ): Болезни аминокислотного обмена; 2) Наследственные нарушения обмена углеводов; 3) Болезни, связанные с
- 5. Болезни аминокислотного обмена Самая многочисленная группа наследственных болезней. К ней относятся: Фенилкетонурия, альбинизм, алкаптонурия и др.
- 6. Наследственные нарушения обмена углеводов Гликогеновая болезнь – нарушение синтеза и разложения гликогена. Развиваются гликогенозы. Болезнь Гирке
- 7. Болезни, связанные с нарушением липидного обмена Болезнь Гоше – связана с дефицитом фермента глюкоцереброзидазы, что приводит
- 8. Женщина - леопард
- 9. Наследственные нарушения обмена стероидов Адреногенитальный синдром (врожденная гиперплазия надпочечников, синдром Уилкинса, сокр. АГС) - Характеризуется повышенной
- 10. Наследственные болезни пуринового и пиримидинового обмена Подагра - заболевание, в основе возникновения которого лежит повышение концентрации
- 11. Болезни нарушения обмена в соединительной ткани Мукополисахаридоз I типа Синдром Марфана Синдром Марфана – мутация в
- 12. Оссифицирующая фибродисплазия - заболеванием, при котором происходит формирование лишних костей на месте синяка или раны. Наследуется
- 13. Синдром Протея - проявляется множественными аномалиями развития (например, частичный гигантизм кистей и стоп, синдактилия, лимфангиомы, липомы,
- 14. Менди Селарс – женщина, страдающая в настоящий момент синдромом Протея.
- 15. Нейрофиброматоз (болезнь Реклингаузена) - аутосомно-доминантное нервно-кожное заболевание с частотой встречаемости 1:2 500 – 4:5 000 (1).
- 16. Наследственные нарушения гема- и порфирина Порфирия или порфириновая болезнь, - почти всегда наследственное нарушение пигментного обмена
- 17. Болезни, связанные с нарушением обмена в эритроцитах Сюда относят болезни, связанные с укорочением жизни эритроцитов, а
- 18. Наследственные нарушения обмена билирубина Синдром Криглера-Найяра - наследуемая негемолитическая желтуха с повышением уровня несвязанного билирубина вследствие
- 19. Жильбера — Мейленграхта синдром (N.A. Gilbert, 1858—1927, франц. врач; E. Meulengracht, р. 1887 г., датский врач;
- 20. Наследственные болезни обмена металлов Гепатоцеребральная дистрофия (болезнь Коновалова-Вильсона) (ГЦД) - наследственное заболевание , характеризующееся поражением нервной
- 21. Гемохроматоз (пигментный цирроз печени, бронзовый диабет синдром Труазье-Ано-Шоффара, сидерофилия и др.) – заболевание, характеризующееся врожденным или
- 22. Наследственные синдромы нарушения всасывания в пищеварительном тракте Муковисцидоз (кистозное перерождение поджелудочной железы, желез кишечника, дыхательных путей)
- 23. Целиакия (болезнь Ги - Гертера - Гейбнера, глютенэнтеропатия, кишечный инфантилизм) — наследственное заболевание, нарушение пищеварения, вызванное
- 24. Хромосомные заболевания Сюда относят болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Они возникают в
- 25. Синдром Дауна (трисомия по 21-й хромосоме) – в 95 % случаев – это типичная трисомия, примерно
- 26. Синдром Эдвардса (трисомия по 18-й хромосоме) – описан Эдвардсом в 1960 г. Девочки болеют чаще в
- 27. Внешний вид больной с синдромом Шерешевского-Тернера Синдром Шерешевского-Тернера – нарушение расхождения половых хромосом. Болеют только женщины,
- 28. Синдром «кошачьего крика» - связан с делецией короткого плеча 5-й хромосомы. Описан Дж. Леженом в 1963
- 29. Болезнь (или синдром) Хантингтона (Хорея Гентингтона) — это заболевание нервной системы, вызванное умножением кодона CAG в
- 30. Болезни, причиной которых является полиплоидия – встречается чаще триплоидия, чем тетраплоидия. Беременность протекает с осложнениями. Основные
- 32. Скачать презентацию

Генные (в основе патологические изменения структуры генов) – 8-10 % смертей
Генные (в основе патологические изменения структуры генов) – 8-10 % смертей

Генные заболевания
1. Чаще моногенные, т.е. в основе патологии изменение одной
Генные заболевания
1. Чаще моногенные, т.е. в основе патологии изменение одной

Классификация генных болезней (ВОЗ):
Болезни аминокислотного обмена;
2) Наследственные нарушения обмена углеводов;
3) Болезни,
Классификация генных болезней (ВОЗ):
Болезни аминокислотного обмена;
2) Наследственные нарушения обмена углеводов;
3) Болезни,

Болезни аминокислотного обмена
Самая многочисленная группа наследственных болезней. К ней
относятся:
Болезни аминокислотного обмена
Самая многочисленная группа наследственных болезней. К ней
относятся:
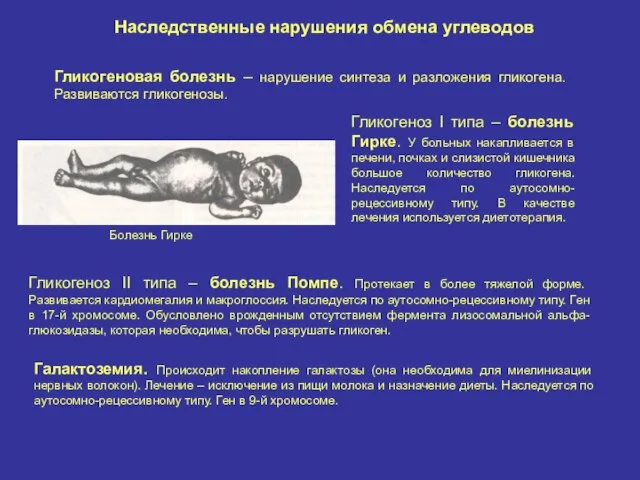
Наследственные нарушения обмена углеводов
Гликогеновая болезнь – нарушение синтеза и разложения гликогена.
Наследственные нарушения обмена углеводов
Гликогеновая болезнь – нарушение синтеза и разложения гликогена.
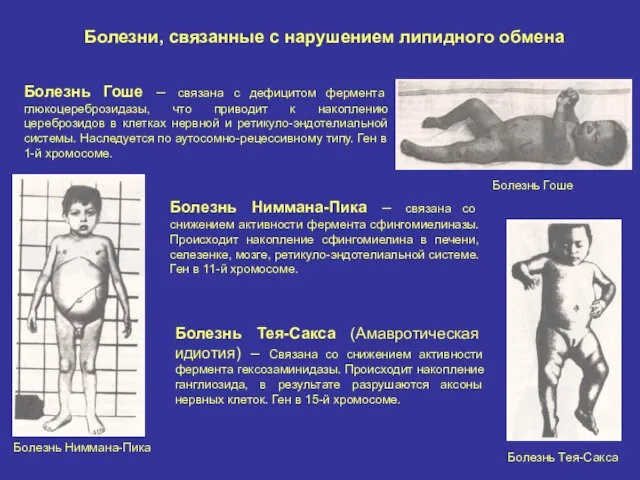
Болезни, связанные с нарушением липидного обмена
Болезнь Гоше – связана с дефицитом
Болезни, связанные с нарушением липидного обмена
Болезнь Гоше – связана с дефицитом

Женщина - леопард
Женщина - леопард

Наследственные нарушения обмена стероидов
Адреногенитальный синдром (врожденная гиперплазия надпочечников, синдром Уилкинса, сокр.
Наследственные нарушения обмена стероидов
Адреногенитальный синдром (врожденная гиперплазия надпочечников, синдром Уилкинса, сокр.

Наследственные болезни пуринового и пиримидинового обмена
Подагра - заболевание, в основе возникновения
Наследственные болезни пуринового и пиримидинового обмена
Подагра - заболевание, в основе возникновения
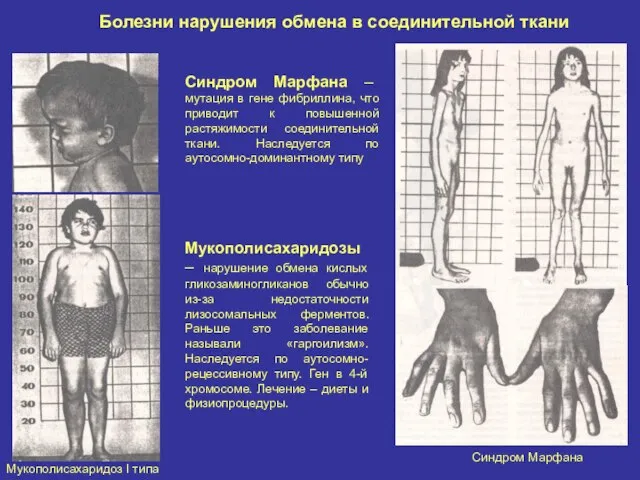
Болезни нарушения обмена в соединительной ткани
Мукополисахаридоз I типа
Синдром Марфана
Синдром Марфана –
Болезни нарушения обмена в соединительной ткани
Мукополисахаридоз I типа
Синдром Марфана
Синдром Марфана –
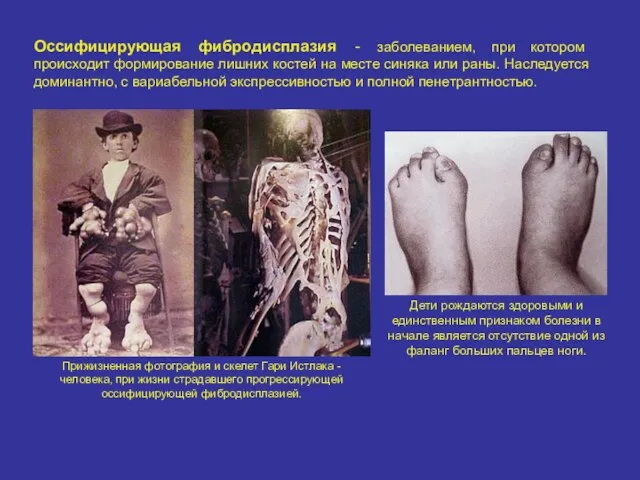
Оссифицирующая фибродисплазия - заболеванием, при котором происходит формирование лишних костей на
Оссифицирующая фибродисплазия - заболеванием, при котором происходит формирование лишних костей на
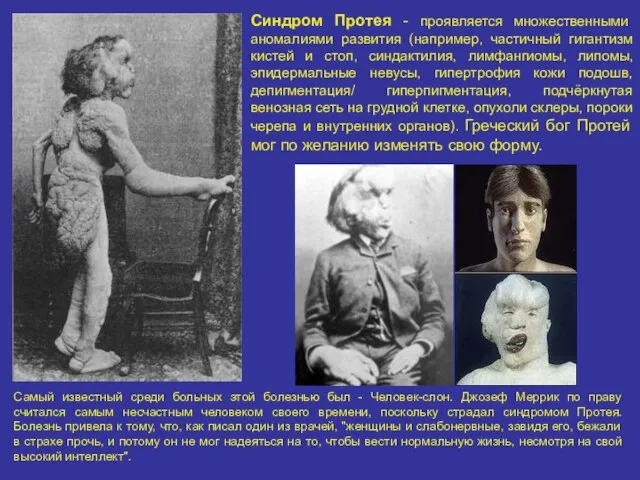
Синдром Протея - проявляется множественными аномалиями развития (например, частичный гигантизм кистей
Синдром Протея - проявляется множественными аномалиями развития (например, частичный гигантизм кистей
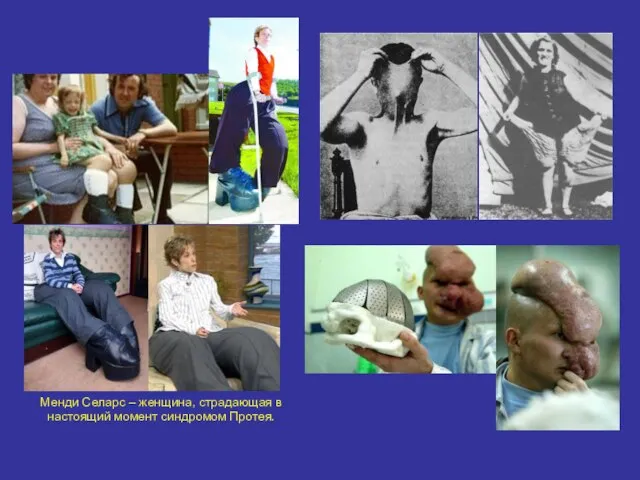
Менди Селарс – женщина, страдающая в настоящий момент синдромом Протея.
Менди Селарс – женщина, страдающая в настоящий момент синдромом Протея.
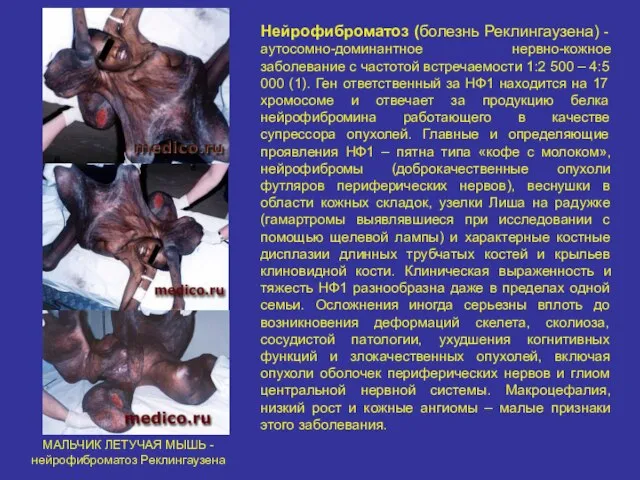
Нейрофиброматоз (болезнь Реклингаузена) - аутосомно-доминантное нервно-кожное заболевание с частотой встречаемости 1:2
Нейрофиброматоз (болезнь Реклингаузена) - аутосомно-доминантное нервно-кожное заболевание с частотой встречаемости 1:2

Наследственные нарушения гема- и порфирина
Порфирия или порфириновая болезнь, - почти всегда
Наследственные нарушения гема- и порфирина
Порфирия или порфириновая болезнь, - почти всегда
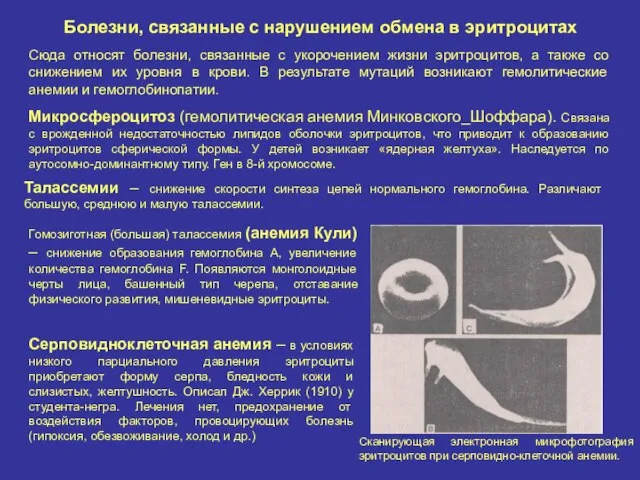
Болезни, связанные с нарушением обмена в эритроцитах
Сюда относят болезни, связанные с
Болезни, связанные с нарушением обмена в эритроцитах
Сюда относят болезни, связанные с

Наследственные нарушения обмена билирубина
Синдром Криглера-Найяра - наследуемая негемолитическая желтуха с повышением
Наследственные нарушения обмена билирубина
Синдром Криглера-Найяра - наследуемая негемолитическая желтуха с повышением

Жильбера — Мейленграхта синдром (N.A. Gilbert, 1858—1927, франц. врач; E. Meulengracht, р. 1887 г., датский врач;
Жильбера — Мейленграхта синдром (N.A. Gilbert, 1858—1927, франц. врач; E. Meulengracht, р. 1887 г., датский врач;

Наследственные болезни обмена металлов
Гепатоцеребральная дистрофия (болезнь Коновалова-Вильсона) (ГЦД) - наследственное
Наследственные болезни обмена металлов
Гепатоцеребральная дистрофия (болезнь Коновалова-Вильсона) (ГЦД) - наследственное

Гемохроматоз (пигментный цирроз печени, бронзовый диабет синдром Труазье-Ано-Шоффара, сидерофилия и др.)
Гемохроматоз (пигментный цирроз печени, бронзовый диабет синдром Труазье-Ано-Шоффара, сидерофилия и др.)

Наследственные синдромы нарушения всасывания в пищеварительном тракте
Муковисцидоз (кистозное перерождение поджелудочной железы,
Наследственные синдромы нарушения всасывания в пищеварительном тракте
Муковисцидоз (кистозное перерождение поджелудочной железы,

Целиакия (болезнь Ги - Гертера - Гейбнера, глютенэнтеропатия, кишечный инфантилизм) —
Целиакия (болезнь Ги - Гертера - Гейбнера, глютенэнтеропатия, кишечный инфантилизм) —

Хромосомные заболевания
Сюда относят болезни, обусловленные геномными мутациями или структурными изменениями
Хромосомные заболевания
Сюда относят болезни, обусловленные геномными мутациями или структурными изменениями
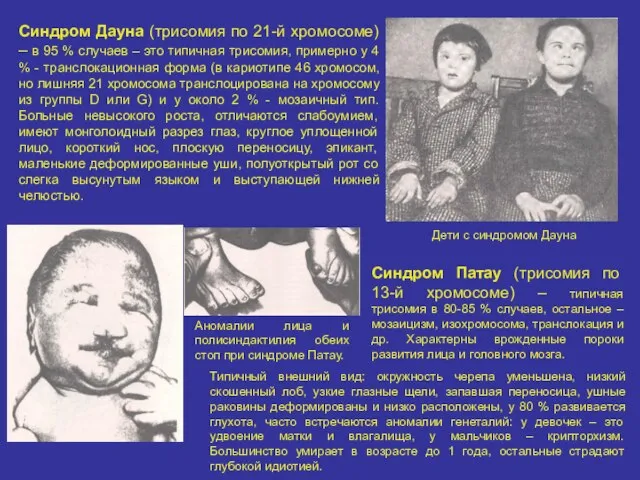
Синдром Дауна (трисомия по 21-й хромосоме) – в 95 % случаев
Синдром Дауна (трисомия по 21-й хромосоме) – в 95 % случаев
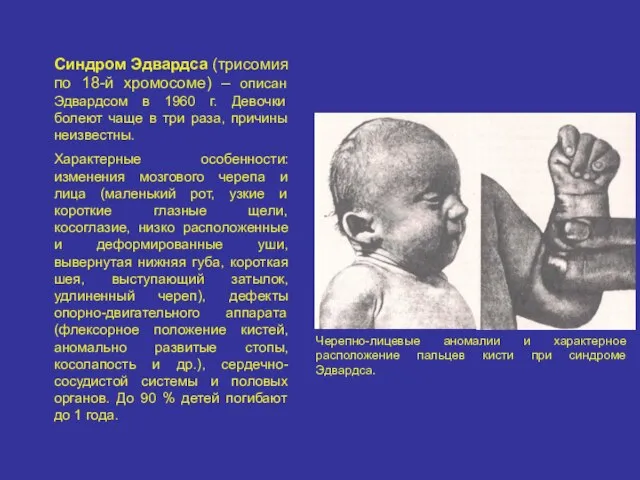
Синдром Эдвардса (трисомия по 18-й хромосоме) – описан Эдвардсом в 1960
Синдром Эдвардса (трисомия по 18-й хромосоме) – описан Эдвардсом в 1960

Внешний вид больной с синдромом Шерешевского-Тернера
Синдром Шерешевского-Тернера – нарушение расхождения половых
Внешний вид больной с синдромом Шерешевского-Тернера
Синдром Шерешевского-Тернера – нарушение расхождения половых
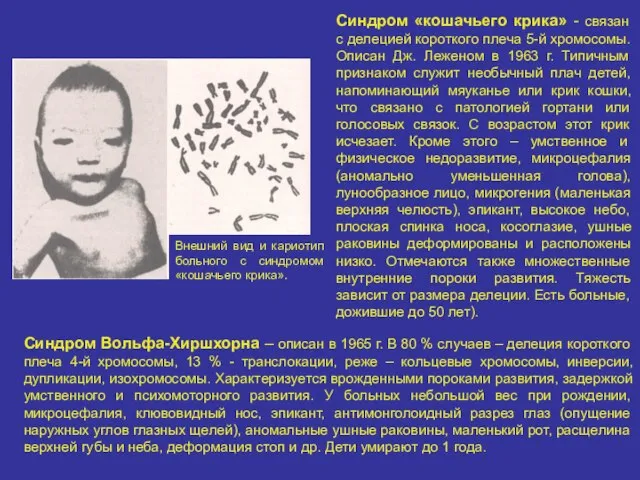
Синдром «кошачьего крика» - связан с делецией короткого плеча 5-й хромосомы.
Синдром «кошачьего крика» - связан с делецией короткого плеча 5-й хромосомы.

Болезнь (или синдром) Хантингтона (Хорея Гентингтона) — это заболевание нервной системы,
Болезнь (или синдром) Хантингтона (Хорея Гентингтона) — это заболевание нервной системы,

Болезни, причиной которых является полиплоидия – встречается чаще триплоидия, чем тетраплоидия.
Болезни, причиной которых является полиплоидия – встречается чаще триплоидия, чем тетраплоидия.
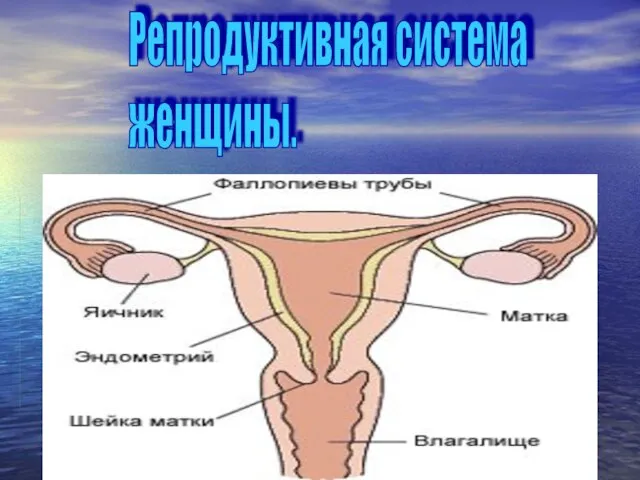 Репродуктивная система женщины
Репродуктивная система женщины Основные заболевания собак
Основные заболевания собак Офтальмология как раздел современной медицинской науки. История офтальмологии и организация офтальмологической службы
Офтальмология как раздел современной медицинской науки. История офтальмологии и организация офтальмологической службы Фондық аурулары бар балаларды бақылау: рахит және рахит тәрізді аурулар
Фондық аурулары бар балаларды бақылау: рахит және рахит тәрізді аурулар Бионические протезы и их роль в жизни человека
Бионические протезы и их роль в жизни человека Процесс становления личности медицинского работника
Процесс становления личности медицинского работника Морфо-функциональные особенности организации сердца. Автоматия
Морфо-функциональные особенности организации сердца. Автоматия Развитие сердца. Основные аномалии сердечно-сосудистой системы. Рентгенанатомия сердца
Развитие сердца. Основные аномалии сердечно-сосудистой системы. Рентгенанатомия сердца Сон и стадии сна
Сон и стадии сна Основы анестезиологии. Местная и общая анестезия
Основы анестезиологии. Местная и общая анестезия Наркотические анальгетики
Наркотические анальгетики Первая помощь при острой сердечной недостаточности
Первая помощь при острой сердечной недостаточности Партограмма – способ графического описания родов
Партограмма – способ графического описания родов Теоретические основы гемостаза и тромбофилии
Теоретические основы гемостаза и тромбофилии Межличностные конфликты, их конструктивное решение
Межличностные конфликты, их конструктивное решение Нарушения проводимости в виде замедления проводимости - блокады сердца
Нарушения проводимости в виде замедления проводимости - блокады сердца Дефекты и осложнения инфузионной терапии
Дефекты и осложнения инфузионной терапии Обеспечение охраны здоровья школьников в условиях распространения COVID-19
Обеспечение охраны здоровья школьников в условиях распространения COVID-19 Өз ойын
Өз ойын Сүйек тіні Сүйек тінінің құрамы: Остеобласттар, Остеоциттер, Остеокласстар
Сүйек тіні Сүйек тінінің құрамы: Остеобласттар, Остеоциттер, Остеокласстар Методы нейровизуализации гидроцефалии и микроцефалии
Методы нейровизуализации гидроцефалии и микроцефалии Кардиогенді шок және жедел коронарлы синдром кезіндегі жедел медициналық көмек көрсету және диагностикалау алгоритмі
Кардиогенді шок және жедел коронарлы синдром кезіндегі жедел медициналық көмек көрсету және диагностикалау алгоритмі Кровотечение и гемостаз
Кровотечение и гемостаз Антибактериальная терапия в педиатрии. Классификация
Антибактериальная терапия в педиатрии. Классификация Типы и уровни интеллектов
Типы и уровни интеллектов Хронический гранулирующий периодонтит. Особенности клинического течения и диагностики. Практическое занятие №5
Хронический гранулирующий периодонтит. Особенности клинического течения и диагностики. Практическое занятие №5 Природное и общественное. Отличия человека от животного
Природное и общественное. Отличия человека от животного Острая кишечная непроходимость
Острая кишечная непроходимость