- Формы ЛВД, ассоциированные с двигательными расстройствами

Содержание
- 2. Клинические подтипы ЛВД: Поведенческая форма ЛВД (пфЛВД) Семантическая форма ППА (сфППА) Аграмматическая форма ППА (афППА) Лобно-височная
- 3. Паркинсонизм Атипичный паркинсонизм – ранняя деменция, частые падения, выраженная вегетативная недостаточность или атаксия. В основе патологического
- 4. Прогрессирующий надъядерный паралич (синдром Стила-Ричардсона-Ольшевского) – спорадическое нейродегенеративное заболевание позднего возраста, для которого характерны: Эпидемиология: распространённость:
- 5. Диагностические критерии ПНП по MDS-PSP (G. Hoglinger, 2017 ): Поддерживающие признаки: Клинические подсказки: (CC1) Отсутствие реакции
- 6. Диагностические критерии ПНП по MDS-PSP (G. Hoglinger, 2017 ):
- 7. Диагностика. у большинства пациентов с ПНП обнаруживаются когнитивные нарушения чаще всего по лобному типу. Наиболее часто
- 8. ЛВД с фенотипом ПНП Наблюдается сочетание ЛВД (поведенческого варианта ЛВД или вариантов ППА) с ПНП-синдромом (как
- 9. Дифференциальный диагноз ЛВД с фенотипом прогрессирующего надъядерного паралича и ПНП
- 10. Кортикобазальный синдром (КБС) Кортикобазальная дегенерация (КБД) – прогрессирующее нейродегенеративное заболевание, поражающее кору лобных и теменных долей
- 11. Диагностические критерии КБС (Armstrong et al., 2013):
- 12. Диагностика. различная степень фокальных или право-/левополушарных когнитивных нарушений с относительно сохранной способностью к обучению и памятью.
- 13. ЛВД с фенотипом КБС Особенности ЛВД-КБС: Развивается вследствие мутации в генах PGRN и C9orf72 афППА иногда
- 14. ЛВД, ассоциированная с болезнью двигательного нейрона (БДН) 2005 г. Mackenzie и соавт.: БДН, ЛВД-БАС и ЛВД
- 15. Прогноз ЛВД, ассоциированная с болезнью двигательного нейрона – 2-3 года ЛВД с фенотипом ПНП – 6-8
- 17. Скачать презентацию

Клинические подтипы ЛВД:
Поведенческая форма ЛВД (пфЛВД)
Семантическая форма ППА (сфППА)
Аграмматическая форма ППА
Клинические подтипы ЛВД:
Поведенческая форма ЛВД (пфЛВД)
Семантическая форма ППА (сфППА)
Аграмматическая форма ППА

Паркинсонизм
Атипичный паркинсонизм – ранняя деменция, частые падения, выраженная вегетативная недостаточность или
Паркинсонизм
Атипичный паркинсонизм – ранняя деменция, частые падения, выраженная вегетативная недостаточность или

Прогрессирующий надъядерный паралич (синдром Стила-Ричардсона-Ольшевского)
– спорадическое нейродегенеративное заболевание позднего возраста,
Прогрессирующий надъядерный паралич (синдром Стила-Ричардсона-Ольшевского)
– спорадическое нейродегенеративное заболевание позднего возраста,

Диагностические критерии ПНП по MDS-PSP
(G. Hoglinger, 2017 ):
Поддерживающие признаки:
Клинические подсказки:
(CC1) Отсутствие
Диагностические критерии ПНП по MDS-PSP
(G. Hoglinger, 2017 ):
Поддерживающие признаки:
Клинические подсказки:
(CC1) Отсутствие
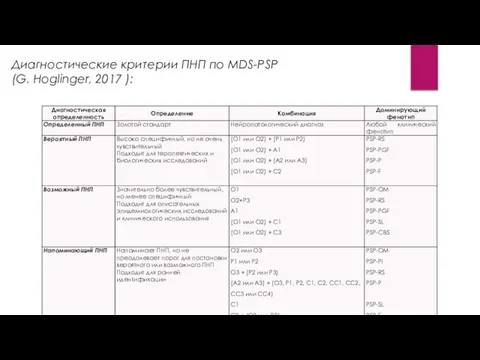
Диагностические критерии ПНП по MDS-PSP
(G. Hoglinger, 2017 ):
Диагностические критерии ПНП по MDS-PSP
(G. Hoglinger, 2017 ):

Диагностика.
у большинства пациентов с ПНП обнаруживаются когнитивные нарушения чаще всего по
Диагностика.
у большинства пациентов с ПНП обнаруживаются когнитивные нарушения чаще всего по

ЛВД с фенотипом ПНП
Наблюдается сочетание ЛВД (поведенческого варианта ЛВД или вариантов
ЛВД с фенотипом ПНП
Наблюдается сочетание ЛВД (поведенческого варианта ЛВД или вариантов
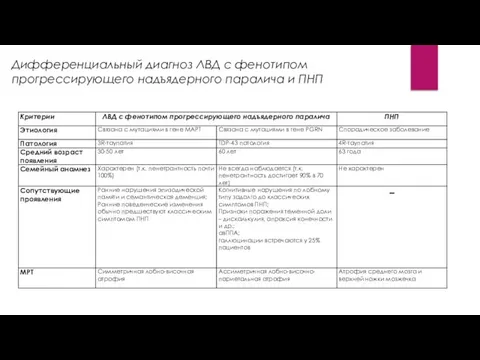
Дифференциальный диагноз ЛВД с фенотипом прогрессирующего надъядерного паралича и ПНП
Дифференциальный диагноз ЛВД с фенотипом прогрессирующего надъядерного паралича и ПНП

Кортикобазальный синдром (КБС)
Кортикобазальная дегенерация (КБД) – прогрессирующее нейродегенеративное заболевание, поражающее кору
Кортикобазальный синдром (КБС)
Кортикобазальная дегенерация (КБД) – прогрессирующее нейродегенеративное заболевание, поражающее кору

Диагностические критерии КБС (Armstrong et al., 2013):
Диагностические критерии КБС (Armstrong et al., 2013):

Диагностика.
различная степень фокальных или право-/левополушарных когнитивных нарушений с относительно сохранной способностью
Диагностика.
различная степень фокальных или право-/левополушарных когнитивных нарушений с относительно сохранной способностью

ЛВД с фенотипом КБС
Особенности ЛВД-КБС:
Развивается вследствие мутации в генах PGRN и
ЛВД с фенотипом КБС
Особенности ЛВД-КБС:
Развивается вследствие мутации в генах PGRN и

ЛВД, ассоциированная с болезнью двигательного нейрона (БДН)
2005 г. Mackenzie и соавт.:
ЛВД, ассоциированная с болезнью двигательного нейрона (БДН)
2005 г. Mackenzie и соавт.:

Прогноз
ЛВД, ассоциированная с болезнью двигательного нейрона – 2-3 года
ЛВД с фенотипом
Прогноз
ЛВД, ассоциированная с болезнью двигательного нейрона – 2-3 года
ЛВД с фенотипом
 Оценка состояния тяжести по системе АВСДЕ (ВОЗ)
Оценка состояния тяжести по системе АВСДЕ (ВОЗ) Гигиена питания
Гигиена питания Жедел және созылмалы жүрек жеткіліксіздігінің патофизиологиясы. Жас балалардағы ерекшеліктері
Жедел және созылмалы жүрек жеткіліксіздігінің патофизиологиясы. Жас балалардағы ерекшеліктері ДЛЯ КОНСУЛЬТАЦИИ (4)
ДЛЯ КОНСУЛЬТАЦИИ (4) Підходи до лікування та вибору препаратів при лихоманці
Підходи до лікування та вибору препаратів при лихоманці Методы анализа белка в биологическом материале
Методы анализа белка в биологическом материале Гипертонический криз
Гипертонический криз Аномалии родовой деятельности. Лечение патологического прелеминарного периода
Аномалии родовой деятельности. Лечение патологического прелеминарного периода Массажер для улучшения циркуляции крови
Массажер для улучшения циркуляции крови Пиелонефрит. Ерте жаста балардағы пиелонефрит
Пиелонефрит. Ерте жаста балардағы пиелонефрит Бронхообструктивный синдром
Бронхообструктивный синдром Рентгенография легких
Рентгенография легких Физиология гемостаза
Физиология гемостаза Осложнения язвенной болезни желудка и двенадцатиперстной кишки
Осложнения язвенной болезни желудка и двенадцатиперстной кишки Инфекционные болезни
Инфекционные болезни Салливан (Sullivan) Гарри Стэк (1892-1949) американский психиатр, психолог, психоаналитик, представитель неофрейдизма
Салливан (Sullivan) Гарри Стэк (1892-1949) американский психиатр, психолог, психоаналитик, представитель неофрейдизма Статьи на тему диагностики (психологическая служба)
Статьи на тему диагностики (психологическая служба) Переломы и вывихи бедра
Переломы и вывихи бедра Боковой амиотрофический склероз
Боковой амиотрофический склероз Физиология. Физиология возбудимых тканей. (Лекция 1)
Физиология. Физиология возбудимых тканей. (Лекция 1) Особенности течения и лечения ХСН у пожилых людей
Особенности течения и лечения ХСН у пожилых людей Балалардағы қалыпты ЭКГ
Балалардағы қалыпты ЭКГ Спонтанный пневмоторакс
Спонтанный пневмоторакс Обеспечения безопасности пищевой продукции
Обеспечения безопасности пищевой продукции Иммунологическая безопасность переливания крови
Иммунологическая безопасность переливания крови Правильное питание – залог здоровья
Правильное питание – залог здоровья Здоровый сон дошкольников. Требования к организации
Здоровый сон дошкольников. Требования к организации Prevention of childhood obesity
Prevention of childhood obesity